TÜRK HEMATOLOJİ DERNEĞİ
HematoLog
2013: 3■2
Dr. Mehmet Sönmez, Dr. Nilay Ermantaş,
Dr. Hasan Mücahit Özbaş
Karadeniz Teknik Üniversitesi Tıp Fakültesi, Hematoloji Bilim Dalı, Trabzon,
Türkiye
e-posta: [email protected]
Anahtar Sözcükler
Lenfoma, İmmun yetersizlik, Tanı ve tedavi
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Özet
Konjenital, edinilmiş veya iyatrojenik nedenlere bağlı olarak immun yetersizlik
gelişen kişilerde lenfoproliferatif hastalıklar normal popülasyona göre daha
sık izlenmektedir. İmmun yetersizlik zemininde oluşan lenfoproliferatif
hastalıklar lenfomalar içerisinde ayrı bir sınıf olarak değerlendirilmektedir.
İmmun yetersizliğin tipi ve şiddetine göre klinik ve patolojik özellikleri
değişkenlik göstermektedir. Ancak hastaların çoğunda gelişen lenfomalar
B lenfositlerden köken alıp agresiv histopatolojik özellikler taşımakta ve
sıklıkla Epstein-Barr virus (EBV) birlikteliği izlenmektedir. Diğer lenfomalara
göre ekstranodal bölge tutulumları daha sık, hastalık progresyonu hızlı,
tedavi yanıtları kötü ve komplikasyon gelişimi daha fazla olmaktadır.
GİRİŞ
Malin hastalıkların oluşumunda birçok faktör etkili olmakla birlikte, tümör
hücrelerinin immün sistem tarafından tanınması ve ortamdan kaldırılmasındaki
yetersizliğinde önemli olduğu bilinmektedir. İmmun yetmezlikli bireylerde
bir hücrenin malin hale dönüşmesi, klonal çoğalması ve kanser oluşumu
daha kolay olabilmektedir. Malin hastalıkların gelişiminde bozulmuş
immün denetimin etkisi, antijenik yapıya sahip olan ve viral enfeksiyonlarda
çoğalabilen hücrelerde daha da belirgin olmaktadır. Bu nedenle, immün
yetersizlikli kişilerde Epstein-Barr virüs (EBV) ile ilişkili lenfoma gelişimi sıklıkla
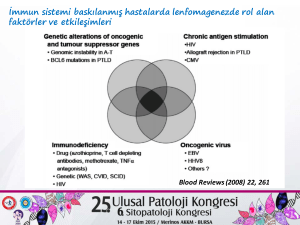
izlenmektedir. Ayrıca onkojenik ve tümör baskılayıcı genlerde kalıtsal veya
sonradan kazanılmış genetik değişiklikler, kronik antijenik uyarım, onkojenik
virüs enfeksiyonları immün yetersizlikli bireylerde lenfoproliferatif hastalık
gelişimine yol açabilen diğer etkenlerdir. Bu etkenlerin birlikteliği ve etkileşimi
512
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Prİmer İmmÜn Yetersİzlİk ZEMİNİNDE Gelİşen
Lenfoprolİferatİf Hastalıklar
Tanım ve Epİdemiyolojİ
Primer immün yetersizlikler (PİY) tekrarlayan ciddi enfeksiyon ataklarıyla
karakterize, otoimmün ve malin hastalıklara eğilimin arttığı heterojen bir
genetik hastalık grubudur. Uluslararası immünoloji dernekleri birliği’nin
2011 yılında yaptığı sınıflamada 175’den fazla PİY tanımlanmıştır. Ancak
tanımlanan hastalıkların çok az kısmında malinensi gelişimi saptanmaktadır.
PİY’ler nadir görülen hastalıklar olup, insidansı 1:10000 olarak belirtilmektedir
(3-5). Hastalarda gelişen malin hastalıklar enfeksiyonlardan sonraki en sık
ölüm nedenidir. PİY’li çocuklarda malin hastalık gelişim riski %4-25 arasında
değişmekte ve malin hastalıkların %60’ını lenfomalar oluşturmaktadır. Aynı
zamanda ileri yaşlarda sağlıklı bireylere göre lenfoma gelişimi 8 kat daha fazla
olarak izlenmektedir. PİY ilişkili lenfomalar çoğunlukla B lenfositlerden köken
almakta ve genellikle difüz büyük B hücreli histolojik tipi görülmektedir. Sıklıkla
merkezi sinir sistemi ve gastrointestinal sistem gibi ekstranodal bölgelerden
kaynaklanmaktadır. Vakaların %30-60’ı EBV ile ilişkili olup, T lenfosit bozukluğu
ile seyreden hastalarda EBV ilişkili lenfomalar daha sık olarak görülmektedir.
Hastalarda izlenen genetik değişiklikler, DNA tamirindeki bozukluklar, diğer
onkojenik virüs enfeksiyonları ve kronik antijenik uyarım lenfoma gelişimi için
diğer risk faktörleri arasında yer almaktadır (1,6,7).
Yaygın Değişken İmmün Yetmezlik (Common Variable İmmün
Yetmezlik-CVİY)
CVİY, B-lenfosit defekti veya B/T lenfosit koordinasyon bozukluğuna bağlı
olarak oluşan, antikor yapımının azalması, immünoglobulin (İg) G, İgA ve/
veya İgM düzeylerinin düşük olması ile karakterize heterojen bir hastalık
grubudur. PİY’ler arasında en sık görülen CVİY genellikle 20-40 yaşlarında
ortaya çıkmakta ve prevalansı 1:25000-50000 arasında değişmektedir (8-10).
Hastalığın klinik bulguları arasında tekrarlayan sinopulmoner enfeksiyonlar,
otoimmün ve granülomatoz hastalık gelişimi yer almaktadır. Yapılan
çalışmalarda CVİY’li hastalarda lenfoma insidansının %1.8-8.2 arasında
olduğu belirtilmektedir (6,9). Hastaların %10’unda ailesel geçiş izlenirken,
%10 hastada tümör nekroz faktörü reseptörü-(TNFRS)13B mutasyonları,
otoimmünite ve lenfoproliferatif hastalıklar ile ilişkilendirilmiştir. Aynı
zamanda, hastalarda izlenen immün bozukluklar, radyosensitivite,
helicobacter pylori, human herpes virus-8 (HHV-8) ve sitomegalovirüs
(CMV) gibi kronik enfeksiyonlar lenfoproliferatif hastalık riskinde artışa yol
TÜRK HEMATOLOJİ DERNEĞİ
lenfoproliferatif hastalık gelişim riskini daha da arttırabilmektedir (1).
İmmün yetersizlik zemininde oluşan lenfoproliferatif hastalıklar Dünya Sağlık
Örgütü (DSÖ) tarafından 4 kategoride sınıflandırılmıştır (2).
1- Primer immün yetersizlik zemininde gelişen lenfoproliferatif hastalıklar
2- İnsan immün yetmezlik virusu (Human immünodeficiency virus, HİV)
infeksiyonu ile ilişkili lenfomalar
3- Post-transplant lenfoproliferatif hastalıklar
4- Iyatrojenik immün yetersizlik sonrası gelişen lenfoproliferatif hastalıklar
513
514
HematoLog
2013:3•2
açmaktadır (6,11). CVİY’de görülen lenfomalar genellikle B hücre kökenli ve
ekstranodal bölgelerde gelişmektedir. Ortalama 40-70 yaşlarında görülür ve
çoğunlukla EBV ile ilişkisizdir (10,11).
Wiskott-Aldrich Sendromu (WAS)
WAS hematopoietik hücrelerde aktin iskeletinin düzenleyicisi olan WAS
proteinini kodlayan aynı adlı genin mutasyonu sonucu oluşan, X’e bağlı geçiş
gösteren, kombine immün yetmezlik ile seyreden bir hastalıktır. Klasik klinik
triadı mikrotrombositopeni, ciddi egzemalar ve tekrarlayan enfeksiyonlardır.
Hastalarda aynı zamanda otoimmün hastalıkların ve hematolojik malinitelerin
sıklığı artmıştır. Doğal öldürücü (NK) hücrelerinin sitotoksik etkilerinde
azalma, EBV gibi onkojenik virüslerin temizlenmesindeki yetersizlik ve
immün denetimdeki bozuklukların hematolojik malinitelerin sıklığında
artışa neden olabileceği düşünülmektedir (6,12). WAS’lı hastalarda NK hücre
fonksiyon bozukluğuyla birlikte, T lenfosit sayı ve fonksiyonunda azalma,
antikor üretiminde yetersizlik ve fagositer, dendritik hücreler, düzenleyici T
hücrelerde çeşitli defektler ve apopitozis artışı gösterilmiştir (5). Hematolojik
malinitelerin büyük çoğunluğunu adölesan ve genç erişkinlerde görülen
lenfoma ve lösemiler oluşturmakta, sıklıkla da EBV ilişkili B-hücreli lenfomalar
görülmektedir (6,13).
X’e Bağlı Lenfoproliferatif Hastalık (XLP)
XLP, SHD1A geni (XLP1) veya BIRC/XIAP geninde (XLP2) mutasyon sonucu
oluşan bir hastalıktır. XLP1 tipi lenfoproliferatif hastalıklarla ilişkili iken, XLP2
tipinde lenfoproliferatif hastalık izlenmemektedir. XLP1 tipinde hemofagositoz
ve karaciğer hasarı ile seyreden şiddetli EBV enfeksiyonu gelişebilmektedir.
SHD1A gen mutasyonu sonucu, T lenfositler antijen sunan B lenfositlerle
etkileşemezken diğer antijen sunan hücrelerle etkileşebildiğinden, bu
hastalarda EBV enfeksiyonlarına yatkınlık ve B hücreli lenfoma sıklığında artış
izlenmektedir. Eşlik eden T ve B lenfositlerin pro-apopitotik özelliklerinin
azalması, NK ve sitotoksik T lenfositlerin immün denetimdeki yetersizlikleri
lenfoma gelişimine yol açabilecek diğer faktörler arasında yer almaktadır
(6,14). XLP1’de hastaların %30’unda ortalama 6 yaş civarında lenfoproliferatif
hastalıklar gelişmektedir. En sık B hücre kökenli yüksek dereceli lenfoma,
özellikle burkitt lenfoma gelişmektedir. Diğer PİY’lerdeki gibi ekstranodal
tutulum sıktır ve sıklıkla ileoçekal bölge tutulumu görülmektedir (6,7).
Otoimmün Lenfoproliferatif Sendrom (OLPS)
OLPS lenfosit apopitoz bozukluğuna bağlı olarak gelişen ve otoimmün
sitopeniler, CD4-CD8 negatif T lenfosit artışı ve lenfoma gelişimine yatkınlıkla
karakterize bir hastalıktır. Bu hastalığı taşıyan kişilerin büyük bir çoğunluğu
otozomal dominant olarak geçiş gösteren FAS geni mutasyonunu heterozigot
olarak taşımaktadırlar. Lenfoma gelişimi 15-20 yaşında olup hodgkin ve B
hücreli hodgkin dışı lenfoma eşit oranda izlenmektedir (6).
Şiddetli Kombine İmmun Yetmezlik (ŞKİY)
ŞKİY, PİY sendromları içerisinde en ciddi klinik bulgularla seyreden, hem
humoral hem hücresel immünite de yetersizliğe yol açan ve birçok farklı
genetik bozukluğu kapsayan bir hastalık grubudur. Tekrarlayan ciddi
enfeksiyonlar nedeniyle tedavisiz yaşam süresi bir yılın altındadır. Yaşayan
hastalarda malinensi görülme oranı %1.5 olup, çoğunluğunu hodgkin dışı
lenfomalar oluşturmaktadır (6,7).
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
AT progresif serebellar ataksi, okülokutanöz telenjiektazi ve disartri ile
karakterize otozomal resesif geçiş gösteren bir hastalıktır. Hastalığın
oluşumundan mutasyona uğramış AT geni sorumludur. AT geni DNA çift
sarmal kırıklarını tespit edip onaran ve hücre siklusunda denetim noktalarının
aktivasyonunu sağlayan bir protein kinazı kodlamaktadır. Bu gendeki
mutasyon progressif nöronal dejenerasyon, hem humoral hem hücresel immün
yetersizlik, radyosensivite ve kanser gelişimde artışa yol açmaktadır. AT PİY’ler
içerisinde en sık kanser gelişimine yol açan hastalıktır. En sık görülen malin
hastalık adölesan dönemde gelişen B-hücreli lenfomalardır (6,7,15).
Kıkırdak Saç Hipoplazisi
Mitokondrial endoribonükleaz işlevinde RNA’yı kodlayan RMRP genindeki
mutasyona bağlı olarak gelişen otosomal resesif geçiş gösteren bir hastalıktır.
Mutasyon sonucu bozulmuş mitokondrial RNA fonksiyonuna bağlı olarak
metafizde kondrodisplazi, seyrek saçlar, anemi, immün yetmezlik ve özellikle
cildi tutan hodgkin dışı lenfoma gelişimi izlenmektedir (6, 7).
Nijmegen Breakage sendromu
BS gen mutasyonuna bağlı olarak kombine immün yetmezlik, kromozomal
instabilite, radyasyona aşırı duyarlılık ve özellikle difüz B hücreli ve
lenfoblastik T hücreli lenfoma gelişiminin eşlik ettiği otozomal resesif bir
hastalıktır (6).
Prognoz ve Tedavİ
PİY zemininde gelişen lenfomaların prognozu gerek tanı anında daha
yaygın hastalık bulgularıyla ortaya çıkmaları, gerekse kemoterapi sonrası
artmış ciddi enfeksiyon riski nedeniyle diğer lenfoma hastalarına göre daha
kötüdür (3). Tedavi yaklaşımı immün yetmezliği olmayan lenfomalı hastalarla
benzerlik göstermekle birlikte, literatürde primer immün yetersizlik ile
birlikte olan lenfomaların tedavi ve prognozu ile ilgili sınırlı sayıda veri
mevcuttur. Bu konuyla ilgili randomize kontrollü çalışma olmadığından
hastaların tedavi planı ve kemoterapi seçiminin, primer immün yetersizliğin
tipi ve prognozuna, gelişen lenfoproliferatif hastalığın histolojik tipine göre
yapılması önerilmektedir (1). İmmunoglobulin eksikliğinde immünoglobulin
replazmanının enfeksiyon sıklığını azalttığı gösterilmiştir (1,3). Hastalarda
Pneumocystis jirovecii gelişimini engellemeye yönelik antibiyotik proflaksisi
yapılmalıdır. Allojenik kök hücre nakli (AKHN) XLP, SKİY ve WAS’lı hastalarda
kür sağlamakla birlikte, morbidite ve mortalitesi yüksektir. Son yıllarda
azaltılmış yoğunluklu rejimler ile başarılı sonuçlar bildirilmektedir (1,16).
İnsan İmmün Yetmezlİk Vİrüs Enfeksİyonu
İle İlİşkİlİ Lenfomalar
Epİdemİyolojİ ve Patogenez
Lenfoma gelişimi insan immün yetmezlik virüs (human immünodeficiency
virus, HİV) enfeksiyonunda morbidite ve mortaliteye yol açan önemli
komplikasyonlardan birisidir (17). Hodgkin dışı lenfoma görülme riski genel
TÜRK HEMATOLOJİ DERNEĞİ
Ataksi-Telenjiektazi (AT)
515
516
HematoLog
2013:3•2
popülasyona göre 60-200 kat artmış olup, her yıl hastaların yaklaşık olarak
%1-6’sında lenfoma gelişmektedir. 1996 yılından sonra HİV enfeksiyonu
tedavisinde kullanılmaya başlanan etkin antiviral tedavilerle hastalarda
lenfoma gelişimi azalmaya başlamıştır (1,18). Ancak önümüzdeki yıllarda,
HİV’li hasta sayısında beklenen artışa bağlı olarak HİV’e bağlı gelişen
lenfomaların giderek artacağı öngörülmektedir (1). Hastalarda en sık görülen
lenfoma tipi difüz büyük B hücreli lenfomadır. Bunu primer merkezi sinir
sistemi lenfoması ve burkitt lenfoma izlemektedir. Daha az sıklıkla primer
efüzyon lenfoması, plazmablastik lenfoma ve hodgkin lenfoma görülmektedir.
Foliküler lenfoma ve periferik T-hücreli lenfomalar ise çok nadir olarak
bildirilmiştir (17,19). Artmış viral yük ve düşük CD4 (+) lenfosit sayısı lenfoma
gelişiminde önemli risk faktörleridir. HİV RNA’sı 100000 kopya/µL’den fazla
ve CD4 (+) lenfosit sayısı 50/µL’den az ise lenfoma gelişme riski belirgin
olarak artmaktadır (18). HİV taşıyan bireylerde kronik antijenik uyarıma bağlı
olarak poliklonal B hücre çoğalması, T lenfositlerin immün denetiminde
bozukluk, immün yetmezlik ile oluşan anormal somatik mutasyonlar ve
bunların üzerine eklenen EBV, HHV-8 gibi viral enfeksiyonların varlığı
lenfoma oluşumunda rol oynamaktadır (1,17,18). HİV’li olgularda gelişen
lenfomalar agressif seyir göstermektedirler. Genellikle tanı anında hastalık
ileri evrede olup, ekstranodal tutulum ve konstitüsyonel semptomlar daha sık
olarak görülmektedir. Ekstranodal tutulum en sık gastrointestinal sistemde
izlenirken, kemik iliği, karaciğer, akciğer, merkezi sinir sistemi, yumuşak
dokular, vücut boşlukları gibi birçok organda tutulum izlenebilmektedir.
Ayrıca hastaların %50’sinde kaposi sarkomu gelişmektedir (17,18,19).
Tanı
İnce iğne biyopsisi çoğunlukla yetersiz olduğundan, tanı için tutulan lenf
nodu veya lezyondan eksizyonel biyopsi gerekmektedir. HİV ilişkili difüz
B hücreli lenfomada histolojik olarak sentroblastik ve immünoblastik
olmak üzere iki alt tip tanımlanmıştır. Sentroblastik tip, yuvarlak yada oval
çekirdekli, çekirdekçiklerin belirgin olduğu büyük lenfoid hücrelerin difüz
tabakalar halinde görülmesi ile karakterizedir. Genellikle CD10 ve bcl-6 gibi
germinal merkez ilişkili belirteçleri taşırlar ve tipik olarak CD20 pozitiftir.
İmmunoblastik varyantta ise %90’dan fazla immünoblast izlenir ve sıklıkla
plazmablastik farklılaşma özellikleri gösterirler, bu durum plazmablastik
lenfomadan ayrımını güçleştirmektedir. Post-germinal merkezden köken
aldıklarından CD10 ekspresyonu izlenmezken, sıklıkla plazma hücre ilişkili
MUM1/IRF4 ve CD138/syndecan-1 ekspresyonu mevcuttur. Mitoz sık izlenir
bu nedenle Kİ67/MIB-1 skoru yüksektir. İmmunoblastik varyantta EBV
birlikteliğinde CD20 ekspresyonu kaybolabilmektedir. CD30, CD38 ve CD71
gibi aktivasyon belirteçleri sıklıkla immünoblastik tipte saptanmaktadır.
Plazmablastik lenfomada neoplastik hücreler, büyük immünoblastik hücreler
ile anaplastik büyük hücreli lenfoma hücrelerine benzer bir morfoloji
gösterebildiğinden, her ne kadar B hücre kökenli olsalar da CD20 ve CD79a
eksprese etmezler. Genellikle CD45, CD30, CD38 ve CD138 ekspresyonu
mevcuttur. Hastalarda aynı zamanda geçirilmiş HHV-8 ve EBV enfeksiyonu
da saptanabilir. Plazmablastik lenfomalar genellikle oral kavite ve çene
bölgesinden köken almakta ve tipik olarak CD38, CD138 ve MUM1/IRF4
ekpresyonu saptanırken, CD20 ve CD45 ekspresyonu izlenmemektedir.
Olguların %50’den fazlasında geçirilmiş EBV enfeksiyonu mevcuttur. HİV
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Evreleme
Diğer lenfomalar gibi hastalar Cotswold tarafından modifiye edilmiş AnnArbor evreleme sistemine göre evrelendirilmektedir. Hastalarda görülebilen
foliküler hiperplazi varlığında pozitron emisyon tomografi yanlış pozitifliklere
neden olabileceğinden, evrelendirilmede tüm vücut tomografi kullanılması
uygun olmaktadır. Bu hastalarda diğer lenfomalardan farklı olarak CD4
pozitif lenfosit sayısı ve HİV viral yüküne bakılması gerekmektedir (18).
Prognoz ve Tedavİ
HİV etkili antiviral tedavilerin kullanımı öncesinde HİV ilişkili lenfomaların
prognozu oldukça kötü ve sağ kalım süresi 5-6 ay ile sınırlı iken, etkin
antiviral tedavilerin kullanıma girmesi ile hastaların %50’sinden fazlasında
uzun süreli hastalıksız sağkalım sağlanmıştır (17,18). 111 HİV (+) agressif
lenfomalı hastada yapılan bir çalışmada uluslararası prognostik skor (IPS)
ve CD4 pozitif hücre sayısının (<100/µl) prognozu öngörmede önemli
olduğu bildirilmiştir (20). Yapılan bir başka çalışmada ise özellikle etoposid,
vinkristin, doksorubisin, prednizolon, siklofosfamid, rituksimab (R-EPOCH)
tedavi rejimi alan hastalarda IPS’nin prognoz üzerine etkisi gösterilememiştir
(17). Merkezi sinir sistemi tutulumu olan hastalarda ise tedavi yanıtları ve
prognoz oldukça kötüdür (17,18).
2005 yılında AIDS malinensileri konsorsuyumu, agressif lenfomaların
tedavisinde kullanılan siklofosfamid, adriamisin, vinkristin, prednisolon
(CHOP) rejimine rituksimab ilave etmekle remisyon oranları artarken CD4
lenfosit sayısındaki azlığa bağlı olarak gelişen enfeksiyonlardan dolayı ölüm
oranlarının da arttığını belirtmiştir (17,21). Takiben yapılan bir başka çalışmada
ise R-CHOP tedavisiyle hastaların %77’sinde tam remisyon izlenirken, 2
yıllık sağkalım %75 olarak bulunmuş ve rituksimab’ın etkin ve güvenli
olduğu bildirilmiştir (22). Sonrasında kemoterapi etkinliği ve tolerabilitenin
arttırılmasına yönelik çalışmalarda R-EPOCH tedavisinin R-CHOP’a göre daha
etkili olduğu izlenmiştir (23). R-EPOCH tedavisi ile 5 yıllık progresyonsuz
sağkalım %84 toplam sağ kalım ise %68 bulunmuş ve hastaların %79’unda 3
kür kemoterapi uygulanmasının yeterli olduğu saptanmıştır (24). Dolayısıyla
HİV(+) difüz büyük B hücreli lenfomaların tedavisinde 2 kür R-EPOCH tedavisi,
sonrası değerlendirmede tam yanıt varlığında 1 kür, parsiyel yanıt varlığında
2-4 kür daha tedavi önerilmektedir. Tedavi sürecinde hastalara nötropenik
dönemlerde G-CSF ve ayrıca pneumocystis jirovecii ve mycobacterium
avium’a yönelik proflaksi uygulanmalıdır. MSS proflaksisi olarak tedavinin 3.
kürünün 1. ve 5. gününde, takiben 3 haftada bir toplam 6 doz intratekal 12
mg metotreksat yeterli olmaktadır. Leptomeningeal tutulumu olan vakalarda
ise intratekal tedavi MSS tutulum bulguları düzeldikten 2 hafta sonrasına
TÜRK HEMATOLOJİ DERNEĞİ
ilişkili burkitt lenfomada ise yüksek mitotik aktivite izlenirken, CD19, CD20,
CD79a ve CD10 ekspresyonu varlığında, bcl-2 negatiftir. Hodgkin lenfoma
gelişen hastalarda ise sıklıkla miks sellüler tip klasik hodgkin lenfoma
görülmektedir. Ancak son yıllarda antiviral tedavilerin kullanıma girmesiyle
nodüler sklerozan tipin görülme oranı artmıştır (17,18).
517
518
HematoLog
2013:3•2
kadar haftada 2 kez (en az 4 hafta süreyle) sonrasında haftada bir, 6 hafta
sonra ayda bir 6 ay süreyle uygulanmalıdır (17). Kemoterapi sürecinde
antiviral tedavinin kemoterapi etkinliğini azaltıcı ve yan etkileri artırıcı etkisi
olmasına rağmen kullanılması uygundur (17,18).
Burkitt veya burkitt benzeri lenfoma gibi hızlı seyirli lenfomalarda
siklofosfamid, vinkristin, doksorubisin, metotreksat, ifosfamid, etoposid
ve sitarabin (modifiye CODOX-M-IVAC) veya benzer ilaçları içeren hyperCVAD (CD20 (+)’liğinde rituksimab ile birlikte) ve R-EPOCH ile oldukca iyi
tedavi yanıtları izlenmiştir. Buna karşılık plazmablastik lenfomalarda tedavi
yanıtları oldukça kötü ve ortalama sağkalım 6 aydan daha kısadır. HİV (-)
hastalardan farklı olarak HİV (+) MSS lenfomalarında yüksek doz metotreksat
ve kombinasyon kemoterapileri etkisiz olup, bu hastalara radyoterapi
planlanmalıdır (7,17,23). Nüks gelişen hastalarda prognoz oldukça kötü
olup, ortalama sağkalım 1 yıldan kısadır. Bu hastalarda kurtarma tedavileri
ardından otolog kök hücre naklinin olumlu sonuç verdiği ve sağkalımı
arttırdığı gösterilmiştir. Ancak allojenik kök hücre naklinin etkinliği ile ilgili
yeterli veri henüz mevcut değildir (17,25).
Tablo 1. PTLH DSÖ 2008 sınıflaması
1- Erken lezyonlar
Plazmasitik hiperplazi
Enfeksiyöz mononükleoz benzeri lezyonlar
2- Polimorfik PTLH
3- Monomorfik PTLH
B hücreli neoplaziler
Difüz büyük B hücreli lenfoma
Burkitt lenfoma
Plazma hücreli miyelom
Plazmasitom benzeri lezyonlar
Diğerleri
T hücreli neoplaziler
Sınıflandırılamayan periferik T hücreli lenfoma
Hepatosplenik T hücreli lenfoma
Diğerleri
4- Klasik hodgkin lenfoma tipi PTLH (HL-PTLH) ve hodgkin benzeri PTLH
İndolent küçük B hücreli lenfomalar PTLH sınıfına dahil edilmemiştir.
Post-Transplant Lenfoprolİferatİf
Hastalıklar
Post-transplant lenfoproliferatif hastalıklar (PTLH), solid organ veya allojenik
kök hücre nakli sonrasında uygulanan immünosupressif tedaviye bağlı olarak
gelişen poliklonal özellik gösteren selim durumlardan, monoklonal karakterde
malin lenfoid proliferasyona kadar değişebilen heterojen bir grup hastalıktır.
Vakaların %70-90’ı EBV reaktivasyonuyla ilişkilidir ve sıklıkla B lenfositlerden
kaynaklanır. Daha az oranda, T ve NK hücrelerinden kaynaklanabilir, bu
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Patogenez
EBV yetişkinlerde yaklaşık %95 sıklıkla orofarengeal dokuda bulunan, herpes
virus ailesinden bir virüstür. Glikoprotein yapıda tabaka ile sarılı EBV genomu
ve nüklear kapsidden oluşur. Primer EBV enfeksiyonu çocukluk çağında
subklinik olarak seyrederken yetişkinlerde ateş, lenfadenopati ve farenjit
ile karakterize enfeksiyöz mononükleoz tablosuna yol açmaktadır. Primer
enfeksiyonda B hücre yüzeyinde bulunan CD21 molekülü EBV glikoprotein
yapının hedef reseptörüdür. Primer enfeksiyon sırasında B lenfositlerin içine
giren EBV genomu B hücrede latent kalan bir episoma dönüşmektedir. EBV
tarafından transformasyona uğratılan ve ölümsüzleştirilen B lenfositlerinin
çoğalması, immün yetmezliği olmayan kişilerde CD4, CD8 T lenfositler ve NK
hücreleri tarafından kontrol altında tutulur. Ancak posttransplant dönemde
uygulanan immünsupressif tedaviler B lenfosit çoğalması üzerindeki bu
kontrolü ortadan kaldırmaktadır. Hücresel düzeyde EBV’ye ait membran
proteinleri, latent membran protein-1(LMP-1) ve LMP-2A antijen aracılı B
hücre aktivasyonunu taklit edebilmektedirler. Ayrıca iki nüklear EBV proteini
EBNA-2 ve EBNA-LP’de c-myc gibi büyüme faktörü öncüllerini uyararak B
hücre çoğalmasına yol açabilmektedir (29,30,31). PTLH’nin yaklaşık %30’unu
oluşturan EBV ilişkisiz hastalığın etyolojisi tamamen aydınlatılamamakla
birlikte, saptanamamış başka virüsler ile diğer kronik antijenik stimulasyon
yapabilen nedenlerin rol oynayabileceği düşünülmektedir. Posttransplant
primer efüzyon lenfoma ise HHV-8 ile ilişkilendirilmiştir (32).
Patolojİk Özellİkler ve Sınıflama
1-Erken lezyonlar: Transplantasyonun ilk yılında gelişen bu tablo çoğunlukla
çocuk ve genç erişkinlerde görülür. Özellikle tonsil ve lenf nodları tutulur
fakat tutulan bölgelerin yapısında bozulma, nekroz gelişimi izlenmez. Malin
transformasyon olmaksızın poliklonal B lenfosit proliferasyonunun olduğu
erken lezyonların, plazma hücre hiperplazisi ve enfeksiyöz mononükleoz
benzeri lezyonların görüldüğü iki subtipi mevcuttur. Plazmasitik hiperplazi
çok sayıda politipik plazma hücreleri ve yer yer immünoblastlardan,
enfeksiyöz mononükleoz benzeri lezyonlar ise plazma hücreleri ve T
lenfositlerle birlikte yer yer immünoblastlardan oluşmaktadır. Bu lezyonlar
spontan veya immünsupressif tedavinin azaltılması ile düzelebilmektedir.
2-Polimorfik PTLH: B hücreli yada T/NK hücreli lenfomaların tüm kriterlerini
karşılamamakla birlikte malin transformasyonu destekleyen poliklonal
yada monoklonal lenfoid infiltratlar izlenmekte ve tüm yaş gruplarında
görülebilmektedir. Lenf nodları yanında gastrointestinal kanal, akciğer ve
allograft tutulumu izlenebilirken, tutulan bölgelerin yapısında bozulma ve
nekroz oluşmaktadır. Immünoblastlar, plazma hücreleri, orta büyüklükte
lenfoid hücreler ve yer yer Reed Sternberg benzeri hücrelerden oluşur. İg
geni ve episomal EBV genomunda klonal patern izlenir.
3-Monomorfik PTLH: Hem B hem de T lenfositlerden kaynaklanmakta,
dokunun yapısı bozulmakta ve diğer organlara yayılım göstermektedir.
B lenfositlerden köken aldığında sıklıkla difüz büyük B hücreli lenfoma ve
TÜRK HEMATOLOJİ DERNEĞİ
durum genellikle EBV ilişkisiz ve kötü prognozla birliktedir. PTLH, DSÖ 2008
sınıflamasında 4 gruba ayrılmıştır. (Tablo 1) (26,27,28).
519
520
HematoLog
2013:3•2
daha az sıklıkla da burkitt lenfoma yada plazmoblastik lenfoma gelişimi
izlenirken, T veya NK hücrelerden kaynaklanması daha azdır. Hemen tüm
vakalarda klonal IgH rearanjmanı ve EBV varlığında epizomal EBV genomu
mevcuttur. Mortalite oranları %80’nin üzerindedir.
4-Klasik hodgkin lenfoma PTLH ve hodgkin lenfoma benzeri PTLH: Patolojik
olarak miks sellüler yada lenfositten fakir hodgkin lenfoma özelliklerini
gösteren nadir bir formdur. Neoplastik hücrelerde CD15 ve CD30 pozitiftir.
CD20 pozitifliği değişken iken CD3 ve CD45 genellikle negatiftir (28).
Sıklık ve Rİsk Faktörlerİ
Transplantasyon sonrası lenfoproliferatif hastalık gelişme riski genel
populasyona göre 30-50 kat artmış olup, transplantasyon yapılan hastaların
%2’sinde görülmektedir. Hastalık gelişimi için en önemli risk faktörleri,
uygulanan immünsupresyonun yoğunluğu ile alıcının nakil öncesi EBV
seropozitiflik durumudur. EBV ile daha önce karşılaşmamış alıcılar yeterli
EBV spesifik T hücre yanıtı oluşturamadıklarından, nakil öncesi EBV’ye
maruz kalmamış olmak hastalık için olumsuz bir etki göstermektedir. Ayrıca
transplantasyonun tipi, hastanın yaşı ve transplantasyon sonrası geçen süre
de hastalığın gelişimini etkilemektedir. Çocukluk çağındaki hastalar genellikle
EBV ile temas etmediklerinden PTLH gelişimine daha yatkındır ve hastalık
transplant sonrası herhangi bir dönemde gelişebilmekle birlikte en sık ilk
bir yılda ortaya çıkmaktadır. PTLH daha çok solid organ nakilleri sonrasında
görülmektedir (Tablo 2). Allojenik kök hücre nakli sonrasında ise, akraba
dışı veya HLA-uyumsuz akraba donörlerden yapılan nakillerde, donörde T
hücre deplesyonu yapıldığında ve spesifik antilenfosit tedavi uygulandığında
hastalık gelişimi artmaktadır. Kronik graft versus host hastalığı geç başlangıçlı
PTLH gelişimine yol açabilmektedir (33,34).
Tablo 2. Transplantasyon tipine göre PTLH sıklığı
Transplatasyon tipi
PTLH
İntestinal ve çoklu organ
Akciğer
Kalp
Karaciğer
Böbrek
Kemik iliği
gelişim oranı
%11-33
%2-9
%2-6
%1%1
<%1
Klİnİk Özellİkler
Hastaların klinik bulguları değişkenlik gösterebilmektedir. Hastalar bazen
asemptomatik olabilirken, bazende ateş, kilo kaybı ve gece terlemesi gibi
nonspesifik yakınmalar ile başvurabilirler. Özellikle çocuk hastalarda daha
sık olmak üzere enfeksiyoz mononükleozu andıran klinik tablo gelişebilir.
Transplant yapılan hastalarda 3 gün ve daha uzun süre devam eden sebepsiz
ateş varlığında PTLH ayırıcı tanıda düşünülmelidir. Klinik bulgular hastalığın
tipine ve tutulum bölgesine bağlı olarak değişkenlik gösterebilmektedir (35,36).
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Hastalarda immünsupressif ilaçları mümkün olan en düşük dozlarda ve
en az kombinasyonla kullanmak hastalığın önlenmesinde etkilidir (37).
Profilaktik ve preemptif antiviral tedavi hastalık insidansını azaltabilmekte
ve gansiklovir asiklovire göre daha belirgin olarak hastalık gelişim riskini
engelleyebilmektedir (38,39). Tedavi yönteminin seçilmesinde hastalık
tipi önemli olmakla birlikte hastalarda öncelikle immünsupressif tedavi
azaltılarak, hastaların doğal immünitesinin onarımı ve EBV ile enfekte B
lenfositlerin proliferasyonunun önlenmesi sağlanmalıdır. İmmunsupressif
tedavinin azaltılması özellikle erken lezyonları olan hastalarda tam
remisyon sağlayabilmektedir. Lokal hastalık varlığında cerrahi yada
radyoterapi uygulanabilir. Retrospektif bir analizde immünsupressif tedavi
azaltılan 30 hasta ile immünsupressif tedavide azaltma ile birlikte cerrahi
yapılan 12 hasta değerlendirilmiş ve kombine tedavide %74 oranında tam
remisyon elde edilmiştir. İmmunsupressif tedavinin azaltılması hastalık
gerilemesini sağlarken, aynı zamanda graft rejekti yada graft versus host
hastalığı gelişimini önleyecek düzeyde olmalıdır. Ancak T hücre hastalığı
immünsupressif tedavi azaltılmasına yanıt vermemektedir (40). Özellikle
polimorfik tipte sık görülen CD20 pozitifliğinde, CD20 monoklonal antikor
rituksimab tedavisi etkilidir. Yanıtsız hastalarda rituksimabın yanına
kemoterapi eklenmesi gerekir. İmmunsupresyon azaltılmasına yanıtsız 43
PTLH’li hastada yapılan bir çalışmada, 4 hafta süreyle 375mg/m²/hafta
rituksimab uygulanmasıyla %44 genel yanıt ve %67 bir yıllık yaşam süresi
sağlanırken, yanıtsız hastalarda rituksimab’a CHOP tedavisinin eklenmesiyle
yanıt oranı %90 olarak saptanmıştır (41,42).
İYatrojenİk İmmÜn Yetmezlİk Zemİnİnde
Gelİşen Lenfoprolİferatİf Hastalıklar
Başta romatolojik hastalıklar olmak üzere otoimmün hastalıkların
tedavisinde kullanılan immünsupressif ilaçlar ile ilişkilendirilen iyatrojenik
immün yetmezliğe bağlı lenfoproliferatif hastalıklar (İİY-LPH), morfolojik
olarak atipik polimorfik lenfoproliferasyon, difüz agressif lenfoma, hodgkin
ve hodgkin benzeri lenfoproliferasyonu içeren heterojen bir gruptur.
Otoimmün hastalıkların tedavisinde kullanılan metotreksat, tiopurinler ve
immünomodülatuar ilaçlar suçlanan ajanları oluşturmaktadır. Romatoid
artrit gibi bazı hastalıkların, tedavi uygulanmasa bile lenfoproliferatif
hastalık riskini arttırıyor olması İİY-LPH gelişiminde neden sonuç ilişkisinin
anlaşılmasını zorlaştırmaktadır. Bundan dolayı lenfoproliferatif hastalık riski
primer hastalığın şiddetiyle de ilişkilendirilmiştir. Ancak, hastalık şiddetiyle
lenfoproliferatif hastalık riskinin artışı, hastalık yanında tedavide daha
yoğun immünosupresif gereksinimine de bağlı olabilmektedir. İİY-LPH’de
özellikle de B hücreli lenfoma ve hodgkin lenfoma gelişmektedir. Etyolojide
suçlanan geçirilmiş EBV enfeksiyonu varlığı İİY-LPH subtipleri arasında
değişkenlik göstermekle birlikte tedaviden bağımsız otoimmün hastalığa
bağlı gelişen lenfoproliferatif hastalıklarda genel olarak izlenmez. EBV
pozitifliği en sık klasik hodgkin lenfomada (%80) görülürken, hepatosplenik
TÜRK HEMATOLOJİ DERNEĞİ
Önleme ve Tedavİ
521
522
HematoLog
2013:3•2
T hücreli lenfomada çoğunlukla saptanmaz. Küçük hasta serilerine dayanan
bilgilere göre son derece nadir bir durum olan İİY-LPH’nin gelişimi için,
kullanılan immünosupressif ilacın tipi, tedavi süresi ve altta yatan hastalık
yanında hastanın cinsiyeti ve lenfoproliferatif hastalığa genetik yatkınlıkta
önem taşımaktadır. Atipik neoplastik olmayan lenfoproliferasyonlar daha
çok erkek hastalarda görülürken, lenfoma gelişimi kadınlarda daha sık
görülmektedir. Hastaların önemli bir bölümü immünsupressif tedavinin
kesilmesiyle kemoterapi yada radyoterapi uygulanmasına gerek olmadan
remisyona girebilmektedir. İmmunsupresif tedavinin kesilmesine yanıt,
gelişen lenfoma tipiyle ilişkili olup, özellikle inflamatuar barsak hastalığında
tiopurin kullanımıyla ilişkilendirilen hepatosplenik T lenfoma immünsupressif
tedavinin kesilmesine yanıt vermemektedir (43,44,45).
Metotreksat
Romatoid artirit tedavisinde uzun süreli ve sık kullanılan metotreksat,
dihidrofolat redüktaz enzimini inhibe ederek DNA sentezinde gerekli
timidin nükleotid yapımını azaltan, sitotoksik etkili antimetabolit bir
ajandır. Romatoid artrit tedavisinde kanser tedavisine göre daha düşük
dozlarda uzun süreli kullanılarak bu hasta grubunda steroid tedavi ihtiyacını
azaltmaktadır. İmmünsupresif etkisini T lenfosit fonksiyonlarını ve T lenfosit
kaynaklı adezyon moleküllerini azaltıp, regülatuar T hücreleri arttırarak
yapmaktadır. Metotreksat kullanımı sonrasında romatoid artritli hastalarda
lenfoproliferatif hastalık gelişim riski 20 kat artabilmektedir. Bu durum
hastalığa, metotreksat kullanımına veya EBV enfeksiyonuna bağlı olarak
oluşabilmektedir. Metotreksat kullanımı sonrası lenfoma ortalama 3 yıl
sonra görülmekle birlikte 10 yıldan daha uzun sürede de gelişebilmektedir.
En sık difüz büyük B hücreli lenfoma, klasik hodgkin ve hodgkin benzeri
lenfoproliferasyon gelişirken, foliküler lenfoma, burkitt lenfoma, periferik
T hücreli lenfoma, lenfoplazmositik lenfoma ve küçük hücreli lenfoma da
görülebilmektedir. Metotreksat tedavisiyle ilişkili lenfomada EBV sıklıkla
saptanmakta ve hastaların %40’ında ekstranodal tutulum izlenmektedir.
Hastaların yaklaşık %20-30’unda tedavinin kesilmesiyle lenfoproliferatif
hastalık gerilemektedir (43,44).
Tiopurinler
Başta Crohn hastalığı olmak üzere inflamatuar barsak hastalıklarının
tedavisinde sık kullanılan tiopurinler (azotipurin, 6-merkaptopurin) 6
tiyoguanine dönüşerek etki gösterir ve nükleotid sentezinin inhibisyonuna yol
açarlar. Sitotoksik T lenfositleri ve NK hücreleri üzerine direkt sitotoksik etki
göstererek immünsupresyon oluştururlar. İnflamatuar barsak hastalıklarında
lenfoproliferatif hastalık riski olmakla birlikte tiopurin kullanımında bu risk 3
ila 5 kat daha artmaktadır (45).
İmmünomodulatuar İlaçlar
Romatoid artrit, psöriasis, ankilozan spondilit, inflamatuar barsak hastalıkları
ve multiple skleroz gibi immünolojik hastalıkların tedavisinde kullanılmak
üzere geliştirilmiş hedefe yönelik tedavilerdir. Tümör nekroz faktör alfa
(TNFα) inhibitörü infliximab, monoklonal antikor adalimumab ve füzyon
proteini etanercept immünmodulatuar ilaçlardan en sık kullanılanlarıdır.
TNFα yolağının inhibisyonu, diğer proinflamatuar sitokinlerde azalmaya
yol açıp stromal ve vasküler mikroçevreyi düzenleyerek antiinflamatuar etki
göstermektedir. Fakat İİY-LPH riskinde artışa hangi mekanizma ile neden
523
İmmün YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
Kaynaklar
1. Tran H, Nourse J, Hall S, Green M, Griffiths L, Gandhi M K. ImmunodeficiencyAssociated Lymphomas. Blood Reviews 2008;22:261-281.
2. Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H et al. WHO Classification of
tumors of haematopoetic and lymphoid tissue. 4th ed. Bosman F. Jaffe E, Lakhani
S, Ohgaki H. editors Lyon, France International Agency for Research on Cancer
(IARC); 2008.
3. Shiramizu B, Wilkinson R, Hayashi R. Lymphoproliferative disorders and
malignancies related to immunodeficiencies. In: Pizzo PA, Poplack DG, editors.
Principles and practice of pediatric oncology. Lippincott Williams & Wilkins; 2011.
4. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients
with primary immunodeficiencies. Anticancer Res 2008;28:1263-1269.
5. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C,
Etzioni A, Fischer A, Franco JL, Geha RS, Hammarström L, Nonoyama S, Notarangelo
LD, Ochs HD, Puck JM, Roifman CM, Seger R, Tang ML. Primary immunodeficiency
diseases: an update on the classification from the International Union of
Immunological Societies Expert Committee for Primary Immunodeficiency.Front
Immun 2011;2:54.
6. Leechawengwongsa E, Shearerb WT. Lymphoma complicating
immunodeficiency syndromes. Curr Opin Hematol 2012;19:305-312.
primary
7. Shapiro RS. Malignancies in the setting of primary immunodeficiency. İmplications
for hematologists/oncologists. Am J Hematol 2011;86:48-55.
8. Salavoura K, Kolialexi A, Tsangarıs G, Mavrou A: Development of Cancer in Patients
with Primary Immunodeficiencies. Anticancer Research 2008;28:1263-1270.
9. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and
mortality in common variable immune deficiency over 4 decades. Blood
2012;119:1650-1657.
10.Cunningham-Rundles C. How I treat common variable immune deficiency. Blood
2010;116:7-15.
11. Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency:
İnterplay between immune dysregulation, infection and genetics. Curr Opin
Hematol 2008;15:368-374.
12.Bosticardo M, Marangoni F, Aiuti A: Recent advances in understanding the
pathophysiology of Wiskott–Aldrich syndrome. Blood 2009;113:6288-6295.
13.Thrasher AJ. New insights into the biology of Wiskott–Aldrich syndrome (WAS).
Hematology Am Soc Hematol Educ Program 2009;132-138.
14.Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, Deenick E, Cook MC,
Riminton DS, Choo S, Loh R, Alvaro F, Booth C, Gaspar HB, Moretta A, Khanna R,
Rickinson AB, Tangye SG. Molecular pathogenesis of EBV susceptibility in XLP as
revealed by analysis of female carriers with heterozygous expression of SAP. PLoS
Biol 2011;9
TÜRK HEMATOLOJİ DERNEĞİ
oldukları bilinmemektedir. Diğer immünmodulatuar ajanlardan daklizumab
CD25 (IL-2 reseptör α zinciri) ve efalizumab ise CD11a (lenfosit fonksiyonuyla
ilişkili antijen 1, LFA-1) inhibisyonuyla otoreaktif T lenfositlerin inhibisyonuna
neden olmaktadır. Romatoid artrit tedavisinde kullanılan anakinra ise IL-1
reseptör antagonisti olarak fonksiyon görmektedir. İmmunomodulatuar
ilaçlarla ilişkili lenfomalarda B hücreli, T hücreli lenfomalar ve hodgkin
lenfoma yada neoplastik olmayan lenfoproliferasyon görülebilmekte ve
tedavinin kesilmesiyle hastalıkta gerileme izlenebilmektedir (46,47).
524
HematoLog
2013:3•2
15.Micol R, Ben Slama L, Suarez F, Le Mignot L, Beauté J, Mahlaoui N, Dubois d’Enghien
C, Laugé A, Hall J, Couturier J, Vallée L, Delobel B, Rivier F, Nguyen K, Billette de
Villemeur T, Stephan JL, Bordigoni P, Bertrand Y, Aladjidi N, Pedespan JM, Thomas
C, Pellier I, Koenig M, Hermine O, Picard C, Moshous D, Neven B, Lanternier F,
Blanche S, Tardieu M, Debré M, Fischer A, Stoppa-Lyonnet D; CEREDIH Network
Investigators. Morbidity and mortality from ataxiatelangiectasia are associated
with ATM genotype. J Allergy Clin Immunol 2011;128:382-389.
16.Cohen JM, Sebire NJ, Harvey J, Gaspar HB, Cathy C, Jones A. et al. Successful
treatment of lymphoproliferative disease complicating primary immunodeficiency
disorders with reducedintensity allogeneic stem-cell transplantation. Blood
2007;110:2209-2214.
17.Dunleavy K, Wilson W H: How I treat HIV-associated lymphoma. Blood Journal
2012;119:3245-3255.
18.Kaplan L D. HIV-associated lymphoma. Best Practice & Research Clinical
Haematology 2012;25:101-117.
19.Longa J L, Engelsa E A, Moorea R D, Geboa K A. Incidence and outcomes of
malignancy in the HAART era in an urban cohort of HIV-infected individuals. AIDS
2008;22:489-496.
20.Bower M, Gazzard B, Mandalia S, Newsom-Davis T, Thirlwell C, Dhillon T, Young
AM, Powles T, Gaya A, Nelson M, Stebbing J. A prognostic index for systemic AIDSrelated non-Hodgkin lymphoma treated in the era of highly active antiretroviral
therapy. Ann Intern Med 2005;143:265.
21.Kaplan LD, Lee JY, Ambinder RF, Sparano JA, Cesarman E, Chadburn A, Levine
AM, Scadden DT. Rituximab does not improve clinical outcome in a randomized
phase 3 trial of CHOP with or without rituximab in patients with HIV-associated
non-Hodgkin lymphoma: AIDS-Malignancies Consortium Trial 010. Blood
2005;106:1538-1543.
22.Boué F, Gabarre J, Gisselbrecht C, Reynes J, Cheret A, Bonnet F, Billaud E, Raphael
M, Lancar R, Costagliola D. Phase II trial of CHOP plus rituximab in patients with
HIVassociated non-Hodgkin’s lymphoma. J Clin Oncol 2006;24:4123-4128.
23.Sparano JA, Lee JY, Kaplan LD, Levine AM, Ramos JC, Ambinder RF, Wachsman
W, Aboulafia D, Noy A, Henry DH, Von Roenn J, Dezube BJ, Remick SC, Shah MH,
Leichman L, Ratner L, Cesarman E, Chadburn A, Mitsuyasu R; AIDS Malignancy
Consortium. Rituximab plus concurrent infusional EPOCH chemotherapy is highly
effective in HIV-associated B-cell non-Hodgkin lymphoma. Blood 2010;115:30083016.
24.Dunleavy K, Little RF, Pittaluga S, Grant N, Wayne AS, Carrasquillo JA, Steinberg
SM, Yarchoan R, Jaffe ES, Wilson WH. The role of tumor histogenesis, FDG-PET, and
shortcourse EPOCH with dose-dense rituximab (SCEPOCH-RR) in HIV-associated
diffuse large B-cell lymphoma. Blood 2010;115:3017-3024.
25.Re A, Michieli M, Casari S, Allione B, Cattaneo C, Rupolo M, Spina M, Manuele
R, Vaccher E, Mazzucato M, Abbruzzese L, Ferremi P, Carosi G, Tirelli U, Rossi
G. High-dose therapy and autologous peripheral blood stem cell transplantation
as salvage treatment for AIDS-related lymphoma: long-term results of the Italian
Cooperative Group on AIDS and Tumors (GICAT) study with analysis of prognostic
factors. Blood 2009;114:1306-1313.
İMMÜN YETERSİZLİK ZEMİNİNDE OLUŞAN
LENFOPROLİFERATİF HASTALIKLAR
525
27.S.Kumar,D.Kumar, D.W.Kingma,E.S.Jaffe.Epstein-Barr virus-associated T
lymphoma in a renal transplant patient. Am J Surg Pathol 1993;1046-1053.
cell
28.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO
classification of lymphoid neoplasms and beyond: evolving concepts and practical
applications.Blood 2011;117:5019-5032.
29.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The
Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the
tumor necrosis factor receptor family. Cell 1995;80:389-399.
30.Liebowitz D. Epstein-Barr virus and a cellular signaling pathway in lymphomas
from immunosuppressed patients. N Engl J Med 1998;338:1413-1421.
31.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The protooncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J
Virol 1999;73:4481-4484.
32.Matsushima AY, Strauchen JA, Lee G, Scigliano E, Hale EE, Weisse MT et al.
Posttransplantation plazmacytic proliferations related to Kaposi’s sarcomaassociated herpesvirus. Am J Surg Pathol 1999;23:1393-1400.
33.Caillard S,Lelong C, Pessione F, Moulin B. French PTLD Working Group. Posttransplant lymphoproliferative disorders occurring after renal transplantation in
adults: report of 230 cases from the French Registry. Am J Transplant 2006;6:27352742.
34.Landgren O, Gilbert ES, Rizzo JD, Socié G, Banks PM, Sobocinski KA et al. Risk
factors for lymphoproliferative disorders after allogeneic hematopoietic cell
transplantation. Blood 2009;113:4992-5001.
35.Penn I. Post-transplant malignancy: The role of ımmunosuppression drug. Saf
2000;23:101-113.
36.Green M. Management of epstein-barr virus-induced posttransplant
lymphoproliferative disease in recipient of solid organ transplantation. Am J
Transplant 2001;1:103-108.
37.Peddi VR,Bryant M, Roy-Chaudhury P, Woodle ES, First MR. Safety, efficacy, and
cost analysis of thymoglobulin induction therapy with intermittent dosing based
on CD3+ lymphocyte counts in kidney and kidney-pancreas transplant recipients.
Transplantation 2002;73:1514-1518.
38.Humar A,Micheals M. American Society of Transplantation recommendation
for screaning, monitoring and reportingof infections complications in
immunsuppression trial in recipient of organ transplantation. Am J Transplant
2006;6:262-274.
39.Funch DP, Walker AM, Schneider G, Ziyadeh NJ, Pescovitz MD. Ganciclovir and
acyclovir reduce the risk of post-transplant lymphoproliferative disorder in renal
transplant recipients. Am J Transplant 2005;5:2894-2900.
TÜRK HEMATOLOJİ DERNEĞİ
26.Kwong YL, Lam CC, Chan TM. Post-transplantation lymphoproliferative disease
of natural killer cell lineage: a clinicopathological and molecular analysis. Br J
Haematol 2000;110:197-202.
526
HematoLog
2013:3•2
40.Reshef R, Vardhanabhuti S, Luskin MR, Heitjan DF, Hadjiliadis D, Goral S, Krok KL,
Goldberg LR, Porter DL, Stadtmauer EA, Tsai DE. Reduction of immunosuppression
as initial therapy for posttransplantation lymphoproliferative disorder. Am J
Transplant 2011;11:336-347.
41.Kalinova L, Indrakova J, Bachleda P. Post-transplant lymphoproliferative disorder.
Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2009;153:251-257.
42.Knight JS, Tsodikov A, Cibrik DM, Ross CW, Kaminski MS, Blayney DW. Lymphoma
after solid organ transplantation: risk, response to therapy, and survival at a
transplantation center. J Clin Oncol 2009;27:3354-3362.
43.Baecklund E, Ekbom A, Sparén P, Feltelius N, Klareskog L. Disease activity and risk
of lymphoma in patients with rheumatoid arthritis: nested case-control study. BMJ
1998;317:180-181.
44.Giard C, Avenel-Audran M, Croué A, Verret JL, Martin L. Primary cutaneous
Epstein-Barr virus-associated B-cell lymphoma arising at the site of subcutaneous
injections of methotrexate. J Clin Oncol 2010;28:717-718.
45.Bewtra M, Lewis JD. Safety profile of IBD: lymphoma risks. Med Clin North Ams
2010;94:93-113.
46.Lee SJ, Chinen J, Kavanaugh A. Immunomodulator therapy: monoclonal antibodies,
fusion proteins, cytokines, and immunoglobulins. J Allergy Clin Immunol
2010;314-323.
47.Hasserjian RP, Chen S, Perkins SL, de Leval L, Kinney MC, Barry TS, Said J, Lim MS,
Finn WG, Medeiros LJ, Harris NL, O’Malley DP. Immunomodulatuar agent related
lymphoproliferative disorders. Mod Pathol 2009;22:1532-1540.