Klasik galaktozemi
Defektif enzim:: Galaktoz-1-fosfat üridiltransferaz (1:40,000)
Mekanizma: Galaktoz-1-fosfat birikimi hücre içi ATP deplesyonuna neden olur (fosforu bağlar); ayrıca
karaciğer, böbrek ve beyin için çok toksiktir.
Klinik bulgular: Semptomlar süt alındıktan birkaç gün sonra başlar ve ilerleyicidir; kusma, ishal, sarılık,
ağır karaciğer fonksiyon bozuklukları, renal tubüler bozukluklar. Ölüm karaciğer ve böbrek yetersizliğine
bağlıdır. Sepsis (özellikle E. Coli) sıktır. Tedavi edilmeyen olgularda bir iki ay sonra bilateral katarakt
(galaktitol birikimi) gelişir.
Tanı: Eritrositlerde galaktoz-1-fosfat üridiltransferaz aktivitesinin düşüklüğü (Beutler testi). Serumda ve
eritrositlerde galaktoz ve galaktoz-1-fosfat (N: 0-0.3 mg/dL) yüksekliği. Renal tübüler hasar: Generalize
aminoasidüri, albüminüri, idrarda glükoz ve galaktoz artışına bağlı indirgeyici madde varlığı (Fehling veya
Benedict testi)
Tedavi: Ömür boyu laktozsuz diyet. Galaktoz-1-fosfat düzeyinin 5mg/dL’nin altında tutulması gerekir.
Komplikasyonlar: Tedaviye rağmen hafif mental retardasyon, ataksi, tremor, gonadal bozukluklar,
ovaryum kistleri
Epimeraz yetersizliği
Olguların çoğu asemptomatik. Semptomatik olgular klasik galaktozemiye benzer.
Galaktokinaz yetersizliği
Klinik bulgular: Hızla ilerleyen bilateral santral katarakt. Böbrek, karaciğer ve beyinde hasar yok.
Tedavide laktozsuz diyet
Kalıtsal früktoz entoleransı
Defektif enzim:: Aldolaz B (1:20,000)
Mekanizma: Früktoz-1-fosfat birikimi hücreiçi ATP deplesyonuna neden olur (fosforu bağlar); ayrıca
karaciğer, böbrek ve beyin için çok toksiktir.
Klinik bulgular: Semptomlar früktozlu gıdaların diyete girmesi ile başlar. Kusma, apati ağır karaciğer
fonksiyon bozuklukları, hepatomegali, hipoglisemi, renal tubüler bozukluklar, kilo kaybı, früktozlu gıdlara
karşı tiksinti, diş çürüğü yok.
Tanı: Früktoz yükleme sırasında bulguların ortaya çıkması (tehlikeli !!!), diyet ile bulguların düzelmesi,
enzim çalışması. Renal tübüler hasar: Generalize aminoasidüri, albüminüri, idrarda glükoz ve galaktoz
artışına bağlı indirgeyici madde varlığı (Fehling veya Benedict testi)
Tedavi: Hayat boyu früktozsuz diyet.
Esansiyel früktozüri
Defektif enzim: Früktokinaz
Klinik bulgular: Yok.
Tanı: İdrarda glükoz dışı redüktan madde (Teşhis tesadüfe bağlı)
Sistinüri
Defekt: Dibazik amino asitlerin(lizin, arjinin, ornitin, ve sistin) renal tübüler reabsorpsiyonunda
azalma
Klinik bulgular: Nefrolitiazis (sistin 1250 µmol/L’nin üzerinde ve asit pH’da kristalleşir)
Tanı: Taze idrarda nitroprussid testi pozitiftir. İdrarda lizin, arjinin, ornitin, ve sistin boşaltımı
artmıştır. Plasma düzeyleri genellikle normaldir.
Tedavi: Fazla (>5L) sıvı alma, idrarın alkalileştirilmesi (üriner enfeksiyonlar !). Seçilmiş olgularda
penisillamin (1-2 g/ gün), merkaptopropionilglisin veya kaptopril.
Lizinürik protein entoleransı
Defekt: Dibazik amino asitlerin (lizin, arjinin, ornitin) intestinal ve renal tübüler reabsorpsiyonunda
azalma üre siklusunu bozar ve lizin yetersizliğine yol açar
Klinik bulgular: Proteinli gıdalara karşı tiksinti, tartı alamama, osteoporoz, hiperammonemi,
ilerleyici ansefalopati.
Tanı: Hiperammonemi, plazmada lizin, arjinin, ornitin düşüklüğü, Yüksek LDH düzeyleri.
Tedavi: Sitrüllin takviyesi, protein kısıtlaması
Hartnup hastalığı
Defekt: Nötral amino asitlerin (alanin, valin, treonin, lösin, izolösin, fenilalanin, tirozin, triptofan, histidin,
glisin) intestinal ve renal tübüler reabsorpsiyonunda azalma; triptofan yetersizliği nikotinik asit (pellegra) ve
serotonin yetersizliğine yol açar.
Klinik bulgular: Fotodermatit, serebellar ataksi; çoğu kez asemptomatik
Tanı: İdrarda nötral amino asitlerin yüksekliği, plazmada düşüklüğü.
Tedavi: Nikotinamid 40-300 mg/gün, güneşten korunma, proteinden zengin diyet
2
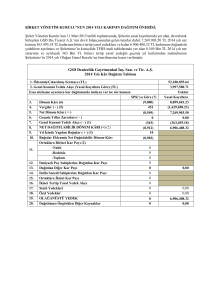
GSD II
GSD IV
alfa-glükosidaz (asit maltaz)
amilo-1,4-1,6 transglükosidaz
GLİKOJEN
Glükoz
Amilopektin
(+) Glükagon
(+) Epinefrin
Fosforilazlar
amilo-1,6
glükosidaz
GSD III
GSD VI
Glükoz
GSD V
EPİMERAZ
YETERSİZLİĞİ
Glikojen sentaz
UDP-glükoz
UDP-galaktoz
Galaktoz-1P üridil transferaz
Glükoz-1-P
KLASİK GALAKTOZEMİ
Galaktoz-1-P
GSD I
GALAKTOKİNAZ
YETERSİZLİĞİ
Glükoz-6-fosfataz
GLÜKOZ
Glükoz-6-P
GALAKTOZ
Galaktitol
Hekzokinaz
- ATP
FRÜKTOZ
Früktoz-6-P
FRÜKTOKİNAZ
YETERSİZLİĞİ
- ATP
FRÜKTOZ-1,6- DİFOSFATAZ
YETERSİZLİĞİ
Fosfofrüktokinaz
GSD VII Früktoz-1,6- dP
Früktoz-1-P
Früktoz-1-P
aldolaz
KALITSAL FRÜKTOZ
ENTOLERANSI
PROTEİN
(+) Glükokortikoid
Gliseraldehit 3-fosfat
+ 2ATP
Gliseraldehit
(+) Glükagon
(+) Glükokortikoid
Gliserol
(+) Epinefrin
(+) Büyüme horm.
(+) Glükokortikoid
(-) İnsülin
Fosfoenolpirüvat (PEP)
Amino asitler
(alanin, glutamat vb)
PEP
+ 2ATP
LAKTAT
KARBOKSİKİNAZ
YETERSİZLİĞİ
PİRÜVAT KARBOKSİLAZ
YETERSİZLİĞİ
(+) İnsülin
KREBS DÖNGÜSÜ
Serbest yağ asitleri
Karnitin
ß-oksidasyon
Asetil CoA
ATP
TRİGLİSERİT
Okzalasetat
Pirüvik asit
PİRÜVAT DEHİDROGENAZ
YETERSİZLİĞİ
GSD 0
Keton cisimleri
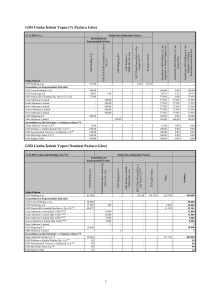
Karbohidrat metabolizmasına genel bakış ve ilgili hastalıklar
Tip
Enzim/sıklık/
kalıtım şekli
Tutulan organlar
Klinik ve laboratuvar
bulguları
Tip 0
(aglikojenoz)
Glikojen sentaz /
nadir/ OR)
Glükoz-6-fosfataz
Tip I a
Liver
/1:100, 000/ OR)
ketozis, hepatomegali (-)
Liver, böbrek,
incebağırsak
(von Gierke)
Neonatal hypoglycemia,
laktik asidoz,ketozis (±},
hepatomegali
Glükoz-6-fosfat transTip I b
Neonatal hypoglycemia,
lokaz /1:1,200,000/ OR
Liver, böbrek,
Leucocyte, intestine
Tip Ia'ya benzer, ayrıca
alyuvar defekti, Crohn hastalığı
Lizosomal alfa
Tip II
glükosidaz/ 1:175,000/
(Pompe)
OR
İskelet ve kalp kası
Hipotoni, dil büyüklüğü,
kardiyomegali, hepato-megali,
vakuollü lenfositler
Tip III
Amilo-1, 6- glükosidaz
(Cori)
/1:125,000/ OR
Liver, iskelet kası
Hipoglisemi (±), ketozis, kas
Karaciğer, iskelet
Tip IV
(Anderson)
Amilo-1, 4-1, 6- trans- kası
tutulması
Hepatomegali, siroz, kas
tutulması
glükosidaz/ 1:1000,000,
OR)
(amilopektinoz)
Kas fosforilazı/
İskelet kası
Halsizlik, kramplar,
1:1000,000,/OR
Karaciğer
Tip V
(McArdle)
Kas fosforilazı/
Hepatomegali, büyüme
1:1000,000/OR
Karaciğer, kan
Tip VI
miyoglobinüri, laktik asidoz
Fosforilaz b-kinaz
gerilği, hipoglisemi(±)
hücreleri
1:125,000, X'R
Hepatomegali, büyüme
gerilği, hipoglisemi(±)
Tip VI a
Fosforilaz b-kinaz
Karaciğer
(nadir, XR)
Hepatomegali, büyüme
geriliği, hipoglisemi(±)
Tip VI b
Fosforilaz b-kinaz
(nadir, OR)
Karaciğer, kan
hücreleri, kas
Hepatomegali, büyüme
geriliği, hipotoni
Tip VI c
Fosforilaz b-kinaz
Kas
(nadir, OR)
Tip VI d
Fosforilaz b-kinaz
Hipotoni
Kalp kası
Glycogen storage disease type I (von Gierke disease)