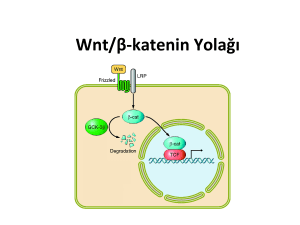
ülseratif kolitte wnt sinyal yolağında rol oynayan bazı genlerin
advertisement

T.C. MERSİN ÜNİVERSİTESİ SAĞLIK BİLİMLERİ ENSTİTÜSÜ TIBBİ BİYOLOJİ ANABİLİM DALI ÜLSERATİF KOLİTTE WNT SİNYAL YOLAĞINDA ROL OYNAYAN BAZI GENLERİN METİLASYONU VE GEN EKSPRESYONU Dr. Zuhal MERT ALTINTAŞ DOKTORA TEZİ DANIŞMAN Prof. Dr. Mehmet Emin ERDAL MERSİN-2012 T.C. MERSİN ÜNİVERSİTESİ SAĞLIK BİLİMLERİ ENSTİTÜSÜ TIBBİ BİYOLOJİ ANABİLİM DALI ÜLSERATİF KOLİTTE WNT SİNYAL YOLAĞINDA ROL OYNAYAN BAZI GENLERİN METİLASYONU VE GEN EKSPRESYONU Dr. Zuhal MERT ALTINTAŞ DOKTORA TEZİ DANIŞMAN Prof. Dr. Mehmet Emin ERDAL Bu tez, Mersin Üniversitesi Bilimsel Araştırma Projeleri Birimi Tarafından BAP-SBE TBG (ZMA) 2009-3 DR numaralı proje olarak desteklenmiştir. Tez No: 25 MERSİN-2012 Mersin Universitesi Saghk Bilimleri Enstitusii Tibbi Biyoloji ve Genetik Anabilim Dali Doktora Programi Cer?evesinde yurutulmus olan Ulseratif Kolitte Wnt Sinyal Yolagmda Rol Oynayan Bazi Genlerin Metilasyonu ve Gen Ekspresyonu basjikh 9ahsma, jurimiz tarafmdan Doktora tezi olarak kabul edilmi§tir. Tez Savunma Tarihil 1/06/2012 Prof. Dr. MehmetVEmin ERDAL Mersin Universitesi Tip Fakultesi Jiiri Ba§kani Prof. Dr. Orhan SEZGlN Prof. Dr. Davut ALPTEKIN Mersirj Universitesf Tip Fakultesi Jiiri Uye Cukurova Universitesi Tip Fakultesi Jiiri Uyesi er BARLAS fversitesi Tip Fakultesi Jtiri Uyesi Bu tez, Enstitii Yonetim Kurulunun Yrd. Do?. DijOzlem IZCI AY Mersin Universitesi Tip Fakultesi Jiiri Uyesi tarih ve.2-Q/2,//D.6. sayih karan ile onaylanmi§tir. Prof. D GLU TEŞEKKÜR Doktora eğitimim boyunca, akademik açıdan yetişmemde büyük katkısı olan, Anabilim Dalı Başkanımız, danışman hocam Sn. Prof. Dr. M. Emin ERDAL’a, tezimin hazırlanması boyunca gösterdiği titizlik, özveri ve bilimsel katkıdan dolayı teşekkür ederim. Anabilim dalımızın değerli hocaları; Sn. Doç. Dr. Nurcan ARAS ATEŞ’e, Sn. Doç. Dr. Etem AKBAŞ’a, Doç. Dr. İ. Ömer BARLAS’a, Yrd. Doç. Dr. Özlem İZCİ AY’a ve Yrd. Doç. Dr. M. Ertan AY’a doktora eğitimim boyunca yaptıkları bilimsel ve akademik katkıdan dolayı teşekkür ederim. Tezimde yer alan hasta gruplarının toplanması sırasındaki yardımlarından dolayı, ME.Ü. Tıp Fakültesi İç Hastalıkları Anabilim Dalı Gastroenteroloji Bilim Dalı Öğretim Üyeleri Sn. Prof. Dr. Orhan SEZGİN’e, Sn. Doç. Dr. Engin ALTINTAŞ’a, Doç. Dr. Fehmi ATEŞ’e, gastroenteroloji asistanlarına ve endoskopi hemşirelerine, tezimin istatistikleri ve bulguların değerlendirilmesi konusundaki yardımlarından dolayı Biyoistatistik ve Tıbbi Bilişim Anabilim Dalı araştırma görevlisi Mehmet Ali SUNGUR’a, eğitimim boyunca, her türlü desteğini esirgemeyen laboratuvarda birlikte çalıştığım arkadaşlarımın hepsine teşekkür ederim. Hayatımın her aşamasında yanımda olan, maddi ve manevi destekleriyle, eğitimimin akademik yönde devam etmesini sağlayan aileme, eşime ve kızlarıma sonsuz teşekkür ederim. iii İÇİNDEKİLER KABUL VE ONAY ii TEŞEKKÜR iii İÇİNDEKİLER iv ŞEKİLLER DİZİNİ vi ÇİZELGELER DİZİNİ vii SİMGELER ve KISALTMALAR DİZİNİ vii ÖZET x ABSTRACT xi 1. GİRİŞ 1 2. GENEL BİLGİLER 4 2.1. Hastalık Aktivitesinin Değerlendirilmesi 4 2.2. Ülseratif Kolit ve Displazi 5 2.3. Malignite Gelişimi 6 2.3.1. ÜK hastalarında kolorektal kanser gelişme riskini etkileyen faktörler 6 2.3.1.1. Kolonik tutulumun yaygınlığı 6 2.3.1.2. Hastalığın süresi 7 2.3.1.3. Hastalığın şiddeti 7 2.3.1.4. Başlangıç yaşı 8 2.3.1.5. Primer sklerozan kolanjit 8 2.3.1.6. Ailede kolorektal kanser bulunması 8 2.4. Kolitle İlişkili Kanserin Klinik ve Patolojik Özellikleri 9 2.5. Patogenez 11 2.6. Wnt/β-katenin sinyal yolağı 16 2.7. Kanserde Wnt/β-katenin sinyali 17 3. GEREÇ ve YÖNTEM 21 3.1. Hasta ve Kontrol Gruplarının Toplanması 21 3.2. RNA İzolasyonu ve RT-PCR 22 3.2.1. Kullanılan Solusyonlar 22 iv 3.2.2. Yöntem 22 3.2.3. Real-Time PCR-Comparative CT (ΔΔCt) Metodu ile Ekspresyon Analizleri 23 3.2.4. Kullanılan Primer ve Problar 23 3.2.5. Real-Time PCR Reaksiyon Ortamının Hazırlanışı 24 3.2.6. Real-Time PCR Reaksiyon Şartları 25 3.2.7. Ekspresyon Tayini 25 3.3. DNA İzolasyonu 26 3.3.1. DNA Modifikasyonu 27 3.3.2. Metilasyon Spesifik PCR İçin Kullanılan Primerler 28 3.3.3. Metilasyon Spesifik PCR Ortamının Hazırlanışı 29 3.3.4. Metilasyon Spesifik PCR Şartları 28 3.3.5. Metilasyon Spesifik PCR Annealing Dereceleri ve Ürün Boyutları 29 3.4. İstatistiksel Analiz 31 4. BULGULAR 32 4.1. Metilasyon Spesifik PCR Bulgularının Değerlendirilmesi 32 4.1.1. SFRP2 33 4.1.2. SFRP4 34 4.1.3. SFRP5 35 4.1.4. APC1 37 4.1.5. APC2 38 4.2. Metilasyon ve İnflamasyon Bulgularının Değerlendirilmesi 39 4.3. Real Time-PCR (Comparative CT) Analiziyle İlgili Bulgular 45 5. TARTIŞMA 52 6. SONUÇLAR ve ÖNERİLER 59 7. KAYNAKLAR 60 8. ÖZGEÇMİŞ 72 v ŞEKİLLER DİZİNİ Şekil 2.1. Kolit ilişkili kolorektal kanserin moleküler patogenezi 12 Şekil 2.2. Sporadik kolorektal kanserin moleküler patogenezi 13 Şekil 2.3. Kanser ve kanser olmayan hücrede Wnt yolağının şematik gösterimi 17 Şekil 2.4. Kanserde Wnt sinyalizasyonunun ilişkisi 19 Şekil 4.1. SFRP2 geninin metile primer ile PCR görüntüsü 33 Şekil 4.2. SFRP2 geninin unmetile primer ile PCR görüntüsü 34 Şekil 4.3. SFRP4 geninin metile primer ile PCR görüntüsü 35 Şekil 4.4. SFRP4 geninin unmetile primer ile PCR görüntüsü 35 Şekil 4.5. SFRP5 geninin metile primer ile PCR görüntüsü. 36 Şekil 4.6. SFRP5 geninin unmetile primer ile PCR görüntüsü 36 Şekil 4.7. APC1 geninin metile primer ile PCR görüntüsü 37 Şekil 4.8. APC1 geninin unmetile primer ile PCR görüntüsü 37 Şekil 4.9. APC2 geninin metile primer ile PCR görüntüsü 38 Şekil 4.10. APC2 geninin unmetile primer ile PCR görüntüsü 39 Şekil 4.11. SFRP2 geninin distal kolon örneklerinin metilasyon ve inflamasyon durumları. 41 Şekil 4.12.SFRP4 geninin distal kolon örneklerinin metilasyon ve inflamasyon durumları. 42 Şekil 4.13. SFRP5 geninin distal kolon örneklerinin metilasyon ve inflamasyon durumları 43 Şekil 4.14. APC1 geninin distal kolon örneklerinin metilasyon ve inflamasyon durumları. 44 Şekil 4.15. APC2 geninin distal kolon örneklerinin metilasyon ve inflamasyon durumları. 45 Şekil 4.16. ACTB genine ait real time PCR amplifikasyon eğrisi 46 Şekil 4.17. SFRP2 genine ait real time PCR amplifikasyon eğrisi 46 Şekil 4.18. SFRP4 genine ait real time PCR amplifikasyon eğrisi 47 Şekil 4.19. SFRP5 genine ait real time PCR amplifikasyon eğrisi 47 Şekil 4.20. APC1 genine ait real time PCR amplifikasyon eğrisi 48 Şekil 4.21. APC2 genine ait real time PCR amplifikasyon eğrisi 48 vi ÇİZELGELER DİZİNİ Çizelge 3.1. Real-Time PCR için kullanılan primer ve problar 24 Çizelge 3.2. Metilasyon Spesifik PCR için kullanılan primerler 28 Çizelge 3.3. Metilasyon Spesifik PCR annealing dereceleri ve ürün boyutları 30 Çizelge 4.1. Hastaların klinik ve inflamatuvar özellikleri 40 Çizelge 4.2. Ülseratif kolit hastaları ile kontrol gruplarının proksimal ve distal kolonlarından alınan örneklerin ekspresyon oranı 50 Çizelge 4.3. Hastaların distal kolon örneklerinde inflamasyon ile genlerinin ekspresyon oranı 51 vii SİMGELER ve KISALTMALAR DİZİNİ ACTB Beta Aktin geni AGPC Asit Guanidinyum-Fenol-Kloroform Yöntemi AOM Azoksimetan APC Adenomatöz polipozis koli Bç Baz çifti C Sitozin CCND1 Siklin D1 CDH1 Kaderin 1 geni cDNA Komplementer Deoksiribonükleik Asit CH Crohn hastalığı CIN Servikal intraepitelyal neoplazi CKI Kazein Kinaz COX Siklooksijenaz CpG Sitozin-Guanin dinükleotidi DCC-DCP4 Kolon ve pankreas kanserinde delesyon olan genler DEPC-H2O Dietilpirokarbonatlı su DKK Dickkopf protein ailesi DNA Deoksiribonükleik asit dNTP Deoksiribonükleotid trifosfat DSS Dekstran sodyum sülfat DVL Disheveled E-CD Epitelyal kaderin G Guanin GSCN Guanidinyum İsotiyosiyonat GSK Glikojen sentez kinaz HMG-box High Mobility Group box hMHL1 İnsan mutL homolog 1 HSK Hepatosellüler kanser İBH İnflamatuvar barsak hastalığı KRK Kolorektal kanser viii Kras Kirsten ras onkogen KCl Potasyum klorür LDH Laktat dehidrogenaz LEF1 Lenfoid arttırıcı bağlayan faktör 1 LOH Heterozigozite kaybı LRP Düşük dansiteli lipoprotein reseptör ilişkili protein l Mikrolitre Min Multiple intestinal neoplazm MSH2 İnsan mutator gen homoloğu MSI Mikrosatellit instabilite MSP Metilasyon Spesifik PCR MYC Traskripsiyon için kodlanan regülatör gen PCR Polimeraz zincir tepkimesi PSK Primer sklerozan kolanjit p14ARF Alternatif okuma çerçevesi p16INK4A Prototipik INK4 protein p53 Tümör protein 53 Real Time PCR Gerçek Zamanlı Polimeraz Zincir Reaksiyonu RNA Ribonükleik asit ROR2 Reseptör-related 2 Rpm Dakikadaki devir sayısı RT-PCR Reverse transkriptaz polimeraz zincir tepkimesi SFRP Çözünür Frizzled ilişkili proteinler TCF T-hücre faktör ÜK Ülseratif kolit WİF WNT inhibitör faktörü ix ÖZET Ülseratif Kolitte Wnt Sinyal Yolağında Rol Oynayan Bazı Genlerin Metilasyonu ve Gen Ekspresyonu Ülseratif kolit; gastrointestinal yolağın kronik inflamatuvar hastalığı olup, kolorektal kanser için artmış riske sahiptir. Ülseratif kolitle ilişkili neoplazide Wnt sinyal yolağının rolü son yayınlarda bildirilmiştir. Bu çalışmada ülseratif kolitle ilişkili neoplazide Wnt sinyal yolağı genlerinin metilasyon ve ekspresyon durumlarının incelemesi amaçlandı. Hasta grubu distal tip ülseratif kolit tanısı almış ve sürveyans kolonoskopisi yapılan 20 bireyden; kontrol grubu kolon kanseri taraması yapılan herhangi bir yakınması ve endoskopik patolojisi olmayan 15 bireyden oluşturuldu. Hasta ve kontrollerin kolonik biyopsilerinden DNA ve RNA elde edildi. SFRP2, SFRP4, SFRP5, APC1, APC2 ve ACTB gen ekspresyonları Real-Time PCR ve “Comparative CT (ΔΔCt)” analizi ile belirlendi. Metilasyon spesifik PCR ile bu genlerin metilasyon durumlarına bakıldı. APC2 geninin metilasyonu için hasta grubunda 8 kişi metile (%40) ve 12 kişi unmetile (%60) iken kontrollerin 1’i metile (%6,7), 14’ü unmetile (%93,3) idi (p=0,018). Hasta grubunda SFRP5 geni için metilasyon durumu ve ekpresyonları arasında anlamlı bir ilişki bulundu (p=0,015). Hasta ve kontroller arasında diğer genler için metilasyon, ekspresyon ve inflamasyon durumu arasında istatistiksel olarak anlamlı ilişki bulunmadı. Ülseratif kolitte APC2’nin metilasyonunun arttığı, SFRP5 metilasyon artışının ekspresyonu azalttığı bulgusunun inflamasyonla ilişkisi olduğu ileri sürülebilir. Ülseratif kolit ilişkili neoplazi ile ilişkisini söyleyebilmek için daha fazla çalışmaya ihtiyaç vardır. Anahtar Kelimeler: Ekspresyon, metilasyon, neoplazi, ülseratif kolit, Wnt Yolağı. x ABSTRACT Methylation and Expression of the Genes that Have a Role in the Wnt Signaling Pathway in Ulcerative Colitis Ulcerative colitis, being chronically inflammatory disease of gastrointestinal pathway, has increased risk of colorectal cancer. The role of WNT signaling pathway in neoplasia related with ulcerative colitis has been indicated in recent publications. In this study, it is aimed to investigate the methylation and expression status of WNT signaling pathway genes in neoplasia related with ulcerative colitis. Patient group composed of 20 people diagnosed with distal type ulcerative colitis and having surveillance colonoscopy, control group composed of 15 people having colon cancer screening, not having any complaints or endoscopic pathology. DNA and RNA are obtained from the biopsies of the patient and control groups. SFRP2, SFRP4, SFRP5, APC1, APC2 and ACTB gene expressions are determined by Real-Time PCR and “Comparative CT (ΔΔCt)” analysis. The methylation status of the genes are studied by using methylation specific PCR. For APC2 gene methylation; in patient group 8 people are methylated (%40) and 12 are unmethylated (%60) whereas in the control group 1 is methylated (%6.7), 14 are unmethylated (93.3%) (p=0.018). In patient group, for SFRP5 gene, a significant relationship is observed between the methylation status and expressions (p=0.015). Regarding the other genes, no significant statistical relationship is observed among the methylation, expression and inflamation status between the patient and control groups. In ulcerative colitis, it is suggested that the findings of the increase in the methylation of APC2, and the decrease in the expression due to the incease of SFRP5 methylation both have a relationship with inflamation. However, further studies are needed in order to suggest its relationship with ulcerative colitis related neoplasia. Keywords: Expression, methylation, neoplasia, ulcerative colitis, WNT pathway. xi 1. GİRİŞ Ülseratif kolitli hastalar kolorektal kanser için artmış riske sahiptir ve ülseratif kolitle ilişkili kolorektal neoplazi bazı hastalarda artmış mortalitenin başlıca nedenidir. Ülseratif kolitle ilişkili neoplazili hastaların prognozunu iyileştirmek için, erken veya prekanseröz evrede tanımak önemlidir. Ülseratif kolitte kolorektal neoplaziye yatkınlık genellikle uzun süren hastalık ve yaygın kolitin varlığı olarak bilinen 2 risk faktörüne bağlıdır (1-3). Ekbom ve ark. (1) kolorektal kanser riski için standardize edilmiş insidans oranlarını proktitli hastalar için 1.7, sol taraf koliti için 2.8 ve total kolit için 14.8 olarak bildirmişlerdir. Son bir meta analizde ülseratif kolitli hastalarda kolorektal kanserin sıklığı 10 yılda %2, 20 yılda %8 ve 30 yılda %18 bulunmuştur (3). Bu nedenle çoklu rastgele biyopsilerle sürveyans kolonoskopi uzun süreli ve yaygın ülseratif kolitli hastalar için önerilmektedir (4-6). Ancak ülseratif kolitle ilişkili neoplaziyi endoskopik olarak saptamak ve histolojik olarak inflamatuvar rejeneratif epitelden ayırt etmek genellikle güç olduğu için ülseratif kolitle ilişkili neoplazinin erken saptanması için geleneksel sürveyans kolonoskopisinin etkili olup olmadığı halen tartışmalı bir konudur. Sürveyans kolonoskopisinin ülseratif kolitle ilişkili neoplazilerin tanısı için yetersiz olması, uzun süreli ve yaygın ülseratif kolitli hastalar arasında neoplazinin artmış riskine sahip kişilerin saptanmasına yardım edecek moleküler belirteçlerin araştırılmasına yol açmıştır. Kanser ilişkili genlerin DNA metilasyon bağımlı sessizleşmesi kolorektal kanserde önemli ve erken bir olaydır. Promotor ilişkili CpG adasının metilasyonu MeCP2, MBD2 ve MBD3 gibi metil bağlama domainlerine sahip çeşitli proteinlerin bağlanmasına yol açar; daha sonra bu proteinler transkripsiyonel sessizleşmeye yol açan olaylar zincirini başlatır (7). Transkripsiyonel sessizleşme histon deasetilazın birikimi yanı sıra histon H3 lizin 9 metilasyonu ve histon H3 lizin 27 metilasyonu gibi histon kuyruklarındaki değişiklikleri de kapsar (8,9). Bu modifikasyonların tümü transkripsiyonel sessizleşmeyi hızlandıran transkripsiyon faktörlerinin girişini kısıtlayan sıkı kompakt kromatine dönüştüren lokal kromatin yapılarındaki değişiklerle sonuçlanır. p16, E-kaderin, hMLH1, p14, HPP1, ER vb gibi genlerin aberan yaş ilişkili kanser spesifik metilasyonu inflamatuvar barsak hastalığı (İBH) ilişkili neoplazi (displazi ve 1 kanserler) durumunda bildirilmiştir (10-14). İBH gibi kronik inflamatuvar durumların tümör süpresör genlerin aberan metilasyonunu ve inaktivasyonunu arttırarak kanserin gelişimini etkileyebileceğine inanılmaktadır. Wnt sinyal yolağı sporadik kolorektal kanser (KRK) gelişiminde önemlidir (1516). Wnt sinyal yolağındaki aktive edici mutasyonlar sporadik KRK’ların %80’ninden fazlasında görülür (17-18). Bir sekrete edilen faktör olan Wnt ligandı sinyaller zincirini başlatacak olan Frizzled (Fz) reseptörüne ve LRP5/6 koreseptörüne bağlandığı zaman Wnt sinyal yolağındaki sinyal başlar (19). Daha sonra Disheveled (Dsh veya dvl) ailesinin proteinleri Fz resptörleri ile etkileşir (20). Takiben bu etkileşimler adenomatöz polipozis koli (APC), AKSİN ve gikojen sentaz kinaz 3β’nin (GSK-3β) kompleks oluşturmasının inhibisyonuna yol açar. Normalde APC/AKSİN/GSK-3β kompleksi β– kateninin fosforilasyonunda ve degradasyonunda rol oynar. APC/AKSİN/GSK-3β kompleksi inhibisyonunun bir sonucu olarak, β katenin MYC, siklooksijenaz 2 (COX-2) ve siklin D1’i içeren birçok kanser ilişkili genlerin transkripsiyonunu nükleusta upregüle eder, fosforillenmemiş β-katenin sitoplazmada birikir ve sonunda nükleusa transloke olur (21-24). Wnt sinyal yolağı gibi kuvvetli gelişimi ayarlayan yolaklar şiddetli negatif regülasyonla kontrol edilir, eğer bu regülasyon bozulursa onların aberran aktivasyonuna ve tümorogenezise yol açabilir. Wnt sinyal yolağı soluble Fz ilişkili proteinler (SFRPs) ve ekstrasellüler olarak Wnt ligandlarına bağlanan Wnt inhibitör faktörü (WIF1) ve düşük dansiteli lipoprotein reseptör ilişkili protein (LRP) yüzey molekülüne bağlanan dickkopf (DKK) ailesinin proteinleri tarafından inhibe edilir (2526). Bu nedenle promotor metilasyonu gibi mekanizmalarla SFRPs, WIF1 ve DKK’lerin inaktivasyonu kanser hücrelerinde Wnt sinyalinin aktivasyonuna yol açabilir. Benzer şekilde mutasyon veya promotor metilasyonu veya her ikisiyle APC’nin inaktivasyonu β kateninin degradasyonunun engellenmesine yol açabilir. Normalde APC daha önce açıklandığı gibi diğer proteinlerle kompleks yaparak βkateninin degradasyonunda rol oynar. Sporadik KRK’larda APC’yi içeren Wnt sinyal yolağı genlerinin mutasyonları bildirilmiş olmasına rağmen bu genlerin mutasyonları İBH ilişkili neoplazilerde (%0-6) nadirdir (27). Ülseratif kolitli hastalar kolorektal kanser için artmış riske sahiptirler ve ülseratif kolitle ilişkili kolorektal neoplazi bazı hastalarda artmış mortalitenin başlıca nedenidir. 2 Ülseratif kolitle ilişkili neoplazili hastaların prognozunu artırmak için erken veya prekanseröz evrede tanımak önemlidir (1-3). Son yayınlarda İBH ilişkili neoplazilerde Wnt sinyal yolağının rolü bildirilmiştir, fakat İBH ilişkili neoplazide bu yolağın epigenetik regülasyonu hakkında çok az şey bilinmektedir (28-30). Artmış nükleer βkatenin ve azalmış stoplazmik APC1A ekspresyonu ülseratif kolit ilişkili kanserlerde bildirilmiştir (31,32). Wnt sinyal yolağı genlerinin mutasyonu İBH ilişkili neoplazilerde nadir bir olay olduğundan Wnt yolağındaki genlerin metilasyonunun bu lezyonlardaki Wnt sinyalinin disregülasyonundan sorumlu olabileceği ileri sürülmektedir. Wnt antogonist genleri tümör supresör olarak işlev gördüğü ve bazı insan malignitelerinin patogenezine katkıda bulunduğu bildirilmiştir. Uzun süreli ÜK’li hastalardan inflame mukozalarında Fz’in artmış ekspresyonu bu reseptörlerin aracılığıyla sinyalin İBH’lı hastalarda malign transformasyonunda ve premalign değişikliklerinde yer alabildiği ileri sürülmektedir. Bu çalışmada Wnt yolağı genlerinin promotor metilasyonunun ve ekspresyonlarının İBH ilişkili karsinogenezisin seyri sırasında ortaya çıkıp çıkmadığını ve ayrıca İBH ilişkili neoplazininin seyri sırasında metilasyon olaylarının ve ekspresyonlarının progresyonu karakterize edip etmeyeceği araştırıldı. Bu İBH ilişkili neoplazilerin erken saptanması için biyomarkırlar olarak araştırmalar için potansiyel adaylar bulmaya yardım edebilir. Bu çalışmada Wnt sinyal yolağı genlerinin metilasyonunun ve ekspresyon durumlarının uzun süredir var olan ülseratif kolitte görülebilen erken olay olup olmadığı ve İBH ilişkili neoplazinin gelişimi sırasında Wnt sinyal yolağı genlerinin metilasyonunda bir artış olup olmadığı ve ekspresyon durumlarını gösterilmesi amaçlanmıştır. 3 2. GENEL BİLGİLER Ülseratif kolit (ÜK) ve Crohn hastalığı (CH); gastrointestinal yolağın kronik inflamatuvar hastalıkları olup, klinik, endoskopik ve histolojik bulguları ile tanımlanırlar. ÜK; etiyolojisi bilinmeyen kronik inflamatuar bağırsak hastalığı grubunda yer alır. Her iki cinsiyeti eşit oranda etkileyebilir (33). Sıklıkla 20-30 veya 70-80 yaş aralığında görülmekle beraber herhangi bir yaşta da ortaya çıkabilir (34,35). Beyaz ırk ve Yahudilerde sık olarak görülen hastalık A.B.D, Güney Avrupa ve Avustralya’da fazla görülmektedir. İnsidans 3-15/100.000, prevalans 80-120/1.000.000 olarak bildirilmektedir. ÜK ilk olarak 1859 yılında Samuel Wilks tarafından tanımlanmıştır. Genellikle rektosigmoid bölgeden başlayarak proksimale doğru ilerler (36). ÜK; kolonda hastalığın tutulum yerine göre sınıflandırılır. Rektum ve sigmoid kolona sınırlı kalırsa ülseratif proktokolit (%40-50), sadece sol kolon tutulumu mevcutsa sol kolit (%30-40), tüm kolon tutulmuşsa pankolit (%20) olarak isimlendirilir (34,37). Hastalık remisyon ve relapslarla karakterize olup rektal kanama, diare ve abdominal ağrı klinik olarak en sık görülen şikâyetlerdir (34). Tanısı ağırlıklı olarak klinik, endoskopik, histopatolojik inceleme ve rezeksiyon materyallerinden konur (38). ÜK’ de görülen mukozal değişiklikler sigmoid kolon ve rektum da belirgin olmakla beraber kolonun diğer lokalizasyonlarına da yayılabilir. İleum, vakaların üçte birinde tutulabilir (34). Bu tutulum yüzeyeldir, çok az klinik önemi vardır ve her zaman kolonik hastalıkla devamlılık gösterir. Sadece rektum ile sınırlı vakalar ise %5-10 oranında görülebilir (37). Apendiks vakaların %20-60’ında, genelde çekum tutulumunun ilerlemiş şekli veya sıçrama lezyon şeklinde görülebilir. Anal lezyonlar %10 vakada saptanır, orta hat fissürleri, deri soyulmaları, akut perianal ve iskiorektal abseler veya rektovajinal fistüllerden oluşabilir (34). 2.1. Hastalık Aktivitesinin Değerlendirilmesi Ülseratif kolitli hastalar, hastalığın lokalizasyonun ve klinik seyrinin yanı sıra; ülseratif kolitin şiddetine göre de sınıflandırılırlar (39). İlk atak esnasında hastaların %50’si hafif hastalığa, %6-19’u da şiddetli hastalığa sahiptirler (40,41). Hastalık 4 aktivitesinin değerlendirilmesi prognozun tahmin edilmesi ve tedavi planlaması için yol göstericidir. Ülseratif kolitte hastalık aktivitesinin değerlendirilmesi için çeşitli yöntemler geliştirilmiştir. Fakat hiçbirisi üniversal bir standart olarak kabul görmemiştir. Bu amaçla en sık kullanılanlardan birisi Truelove-Witts’in sınıflamasıdır (42,43). Truelove-Witts kriterlerine göre hastalığın klinik şiddeti; klinik ve laboratuar bulgularına göre hafif, orta ve ağır olarak sınıflandırılır. (42,43). Burada; dışkılama sıklığı, rektal kanama, ateş, taşikardi, anemi ve sedimantasyon hızına göre sınıflandırma yapılır. Truelove-Witts sınıflaması klinik pratikte kullanım için güvenilir ve basit bir sınıflamadır. Yaygın kolitli hastalarda kullanım için çok uygun olmasına karşın sınırlı hastalığı olan hastalarda hastalık şiddetinin değerlendirilmesi için yeterli değildir. Truelove-Witts sınıflaması hastalık şiddetinin değerlendirilmesi için endoskopik bulguları dikkate almaz (43). Ayrıca kolitin ciddiyet derecesini endoskopik görünüme dayandıran değerlendirme yöntemleri varsa da tedavi edici karar endoskopik görünüm değil öncelikle klinik duruma dayandırılmalıdır (36). Sınırlı hastalığı olanlarda ve klinik araştırmalarda, daha yararlı olan bir sayısal aktivite değerlendirme yöntemi “Ülseratif kolit hastalık aktivite indeksi” dir (43). Bu indeks 4 komponentten elde edilen skorların toplanması ile hesaplanır. Bu komponentler: dışkılama sayısı, rektal kanama, sigmoideskopik bulgular ve hekimin genel değerlendirmesinden oluşur. Bu hastalık aktivite indeksinde elde edilen skor 0-12 arasında değişir. Genel olarak 0-2 puan alanlar remisyonda kabul edilir. Eğer skor 10’dan fazla ise hastalığın şiddetli olduğunu gösterir. Daha yüksek skorlar daha şiddetli hastalığı gösterir. Başlangıç skoruna göre 3 puan ve ya daha fazla azalma tedaviye klinik cevap olarak kabul edilir (43). 2.2. Ülseratif Kolit ve Displazi Displazi, intestinal epitelin neoplastik değişimi olarak tanımlanır. Makroskopik olarak düz, plak, nodül ya da adenoma benzer polipoid yapılanmaları içeren mukozada saptanır. Histolojik olarak displazi tanısı; hücresel ve nükleer pleomorfizm, nükleer hiperkromazi, nükleer polarite kaybı, belirgin nükleer tabakalanma artışı ile konulmaktadır (37). Normalde akut kolitin tamir sürecinde displazi ile reperatif atipiyi (tamirsel değişiklikleri) karıştırmak mümkündür. Özellikle ÜK’li hastalarda devam eden inflamasyon ve reperatif etkiler nedeniyle displazi tanısı koymak zordur. Reperatif 5 etkilerde görülen kriptlerde dallanma, neoplastik proliferasyonla çok rahat bir şekilde karışabilir. Kural olarak akut inflamasyonlu alanlarda displazi tanısı verilmemelidir (34). ÜK hastalarında displaziyi bulma ve evreleme sonuçları tecrübeli patologlar arasında bile değişiklikler göstermektedir (44). Displazinin varlığı arasındaki uzlaşma %68-84 arasında değişmektedir. Atipi varlığı %90’dan fazla oranda, yüksek dereceli displazinin varlığı ise %33-100 arasında değişiklik göstermektedir (45). Kronik kolitler de displazi Rindell’in modifiye edilmiş klasifikasyonuna göre; Displazi olmayan grup: Normal mukoza, inaktif kolit, aktif kolit Displazi belirsiz grup: Muhtemel negatif, bilinmeyen, muhtemel pozitif Displazi mevcut grup: Düşük dereceli ve yüksek dereceli olarak ayrılmaktadır (37). 2.3. Malignite Gelişimi Kronik ülseratif kolitli hastalarda artmış kolon kanseri gelişme riski vardır. Malignite riski ülseratif kolitin en korkulan yönüdür (36,46). Sporadik kolon kanserine zıt olarak ülseratif kolitli hastalarda kanserin adenomatöz polipten menşe alması gerekmez. Fakat kolonoskopide yanındaki displastik olmayan mukozadan ayırt edilemeyen düz displastik epitelyumdan menşe alabilir. Ülseratif kolitli olmayan hastalar gibi ülseratif kolitli hastalarda displastik değişiklikler gösteren polipler gelişebilir (36). 2.3.1. ÜK hastalarında kolorektal kanser gelişme riskini etkileyen faktörler: 2.3.1.1. Kolonik tutulumun yaygınlığı: Ülseratif proktitli hastalarda risk; uygun yaş grubu kontrollere benzerken, hastalığı splenik fleksura proksimaline yayılan hastalar daha yüksek risk taşır. Sol taraf kolitli hastaların orta bir risk taşıdığı var sayılır (36). Pankolonik ülseratif kolit hastalarında kolorektal kanser riski: Dünyadaki değişik merkezlerden elde edilen, 20. yıldan fazla süren ülseratif kolitli hastaların kümülatif kanser insidans oranları %3,1 ile %16 arasında değişmektedir. 30. yılda ise kümülatif kanser riski %30’dur (47-51). Bu farklılık kolektomi oranlarındaki farklılıktan kaynaklandığı düşünülmektedir. Kanada, Winnipeg’de yapılan toplumsal temelli çalışmada İBH’lı hastalar aynı yaşta, cinsiyette, aynı yılda ve aynı yerde yaşayan insanlardan oluşan bir toplum grubu ile karşılaştırılmıştır. Bu çalışmadaki ülseratif kolitli hasta sayısı 2672 idi. Bu çalışmada ülseratif kolitli hastalarda, kolorektal kanser 6 insidansı için relatif risk 2.75 olarak bulunmuştur (52). Ülseratif kolitli hastalarda, kolorektal kanser insidansı için relatif risk, İsveç’te yapılan bir toplumsal çalışmada 5,7: (53, 43), Danimarka çalışmasında ise 1.8 olarak bulunmuştur (1,3,54). Ülseratif kolitte kolon kanseri riskinin araştırıldığı bütün çalışmaların incelendiği bir meta-analizde kümülatif kolorektal kanser insidansı 10. yılda %2, 20. yılda %8, 30. yılda ise %18 olarak saptanmıştır (3). Sol tarafı tutan ülseratif kolit hastalarında kolorektal kanser riski: Eldeki verilere göre proktit ve proktokoliti olan ülseratif kolitli hastalarda kolorektal kanser insidansında belirgin bir artış yoktur (2). Yapılan kontrollü çalışmalarda sol kolonu tutan vakalarda 20. yılda kolorektal kanser insidansı %3.8-%5 arasında değişmektedir. Bu veriler sonucunda sol taraf koliti olan hastalarda kolorektal kanser için ortalama bir risk artışı olduğu düşünülmektedir. Gilat ve ark. (55) 20. yılda kümülatif kanser insidansını proktitli hastalarda sıfır, sol kolonu tutan hastalarda ise %3.8 olarak saptamıştır. Bütün bu çalışmalar sol taraf koliti olanlarda kolon kanseri riskinin pankolit olanların üçte bir ila dörtte biri kadar olduğunu göstermektedir. 2.3.1.2. Hastalığın süresi: Çalışmalarda riskin büyüklüğü farklı ise de bütün çalışmalarda uzun devam eden ülseratif kolitli hastalarda, kolon kanseri riski yaş uyumlu nüfusa göre anlamlı olarak yüksektir (36). Hastalığın süresi 8 yıldan daha kısa ise kolorektal kanser ile çok nadiren karşılaşılır. Daha sonraki yıllarda kanser gelişme riski her yıl %0.5 ila %1 arasında artar (46). Tersiyer referans merkezlerinde yapılan çalışmalarda bildirilen kanser riski 20 yıldan sonra %13, 30 yıldan sonra %34 iken, nüfusa dayalı çalışmalarda risk 20 ve 30 sene sonra sırasıyla %5.5 ve %13’tür (36). Eldeki veriler, pankolitli hastalarda 8. yıla kadar, sol taraf koliti olan vakalarda ise 12 yıla kadar kanser için tarama çalışmalarına başlamaya gerek olmadığını göstermektedir (46). 2.3.1.3. Hastalığın şiddeti: Daha şiddetli inflamasyonun, daha çok genetik mutasyona ve artmış kanser riskine yol açtığına dair elde güçlü veriler yoktur. Ancak daha şiddetli hastalarda kolektomi olma ihtimalinin daha yüksek olması nedeniyle şiddetli aktiviteli hastalarda kolerektal kanser gelişme riski yapay olarak düşecektir (46). 7 2.3.1.4. Başlangıç yaşı: İsveç’te yapılan toplum temelli iki araştırmada erken yaşta teşhis edilen hastalarda artmış bir kanser riski olduğu gösterilmiştir (1,56). Karlen ve ark. (56) 1955-1989 yılları arasında 1547 ülseratif kolitli hastada 29 yaşının altındakilerde relatif riskin %3.0 (CI= %95) olmasına karşın daha yaşlı grupta relatif risk %2.0-4.2 olduğunu göstermişlerdir. Benzer şekilde Ekbom ve ark. (1) 14 yaşından daha genç ülseratif kolitli hastaların en yüksek riske sahip olduğunu göstermişlerdir. “Multivariate” analizler başlangıç yaşının kolorektal kanser için bağımsız bir risk faktörü olduğunu göstermektedir. Danimarka çalışmasında başlangıcı 0-19 yaşları arasında olanlarda kolorektal kanser gelişme için relatif risk 163 iken; başlangıcı 20-39 yaşları arasında olanlarda relatif risk 22; Kanada’daki bir toplumsal temelli çalışmada, 40 yaşından önce tanı konulan vakalarda artmış kolon kanseri insidansı için en yüksek risk (relatif risk 12.4) saptanmıştır. Ancak tüm çalışmalarda kolorektal kanser riski ile erken yaşta teşhis arasındaki ilişki gösterilememiştir (46). Bu konuda yeni çalışmalara ihtiyaç vardır. 2.3.1.5. Primer sklerozan kolanjit: Ülseratif kolitli hastalarda primer sklerozan kolanjit (PSK) insidansı yaklaşık olarak %5’dir. Çeşitli vaka serileri ÜK ve PSK’li hastaların, yalnız başına ÜK olan hastalara göre kolorektal kanser için yüksek risk altında olduklarını göstermektedir. Cleveland Klinik’te 1976-1994 yılları arasındaki PSK’si olan 132 ÜK hastasında kolorektal kanser gelişme riski %13 iken, PSK’si olmayan ÜK hastalarda kanser gelişme oranı %3 olarak bulunmuştur. Kanser ve displazi için relatif risk 3.2 idi (57). İsveç’te ki bir toplum temelli çalışmada PSK’i olan 104 ÜK hastasında kümülatif kanser gelişimi oranları teşhisten sonraki 10. yılda %10, 20. yılda %33, 30. yılda ise %40 olarak bulunmuştur (46). 2.3.1.6. Ailede kolorektal kanser bulunması: Birinci derecede akrabaları arasında kolorektal kanser bulunanlarda kolorektal kanser gelişme insidansı yaklaşık olarak iki kat daha yüksektir (58, 59). İsveç’te yapılan toplum temelli bir çalışmada 1941 ile 1995 yılları arasında takip edilen 19.876 kişide birinci derecede akrabalarında kolorektal kanser bulunan ÜK hastalarında, birinci derecede akrabalarında kolorektal kanser olmayan ÜK hastalarına göre relatif risk 2.5 olarak bulunmuştur. Bu grup içinde birinci 8 derecede akrabalarında kolorektal kanser gelişimi 50 yaşından önce olanlarda relatif risk 9,2 olarak bulunmuştur (46). ÜK veya Crohn kolitli İBH hastaları KRK gelişimi için artmış riske sahiptir. Bu riskteki artış genetik bir yatkınlıktan daha çok gastrointestinal mukozanın kronik inflamasyonu sonucudur. Çoğu KRK’lar genelde displastik öncü bir lezyondan gelişir. Sporadik KRK’larda displastik öncü genellikle endoskopik polipektomi ile tipik olarak uzaklaştırılan neoplazinin ayrı bir odağı olan adenomatöz polip (adenoma)’dır. Aksine polipoid, düz, lokalize veya multifokal displastik lezyon gelişen İBH’lı hastalar kolon inflamasyonunun işaretidir ve neoplazi için artmış bir risktir ve sıklıkla tüm kolon ve rektumun cerrahi çıkarımı için gerekli endikasyondur. Lezyonların biyolojik davranışı ve morfolojisindeki bu farklılıklar sadece İBH’lı hastalar arasında KRK taraması için dürtü oluşturmaz aynı zamanda kronik inflamasyonun KRK’nın gelişimine nasıl katkıda bulunduğunun sorusuna yol açar. Sporadik kolorektal tümörlerin gelişimi sırasında ortaya çıkan adenomadan karsinomaya ilerleme İBH ile ilişkili KRK’da inflamasyondisplazi-karsinom sırasını gösterebilir (60). 2.4. Kolitle İlişkili Kanserin Klinik ve Patolojik Özellikleri Kronik inflamasyonun İBH’lı hastalarda KRK için bir anahtar risk faktörü olduğunu gösteren birkaç kanıt dizisi bildirilmiştir (61, 62). Gelişen KRK için risk, kolitin daha uzun süresi ile artar. KRK’ya 7 yıldan daha az koliti olan kişilerde nadir rastlanırken; daha sonra risk her yıl için yaklaşık olarak %0,5-1 olarak artar. Kolitin yaygınlığı diğer bir risk faktörüdür, kolitle tutulu daha fazla kolonik yüzey kolon kanseri için daha büyük bir risktir. Ancak rektuma sınırlı inflamasyonu olan hastalar kanser riskinde anlamlı artışa sahip değildirler. KRK riski safra kanalı kanserine de yatkınlığı olan safra kanalının kronik inflamasyonu ile karakterize bir idiopatik durum olan primer sklerozan kolanjiti de olan İBH’lı küçük hasta grubunda da oldukça yüksektir (60). Bazı çalışmalar antiinflamatuvar ilaçlar özellikle mesalamin ve muhtemelen kortikosteroidler İBH’lı hastalar arasında kolorektal displazi ve kanser gelişme olasılığını azaltığını bildirmiştir. Nonsteroid antiinflamatuvar ilaç alan sağlıklı kişiler ve familyal adenomatöz polipozisli hastalar için adenom boyut ve sayısının azaldığına ait benzer bulgular bildirilmiştir (60). 9 KRK patogenezinde kronik inflamasyonu gösteren kanıtlara rağmen birkaç çalışmada İBH’lı hastalarda KRK riski ile inflamasyonun düzeyi arasında bir korelasyon olup olmadığını doğrudan araştırılmıştır. Semptomatik alevlenmenin sıklığına göre ölçülmüş hastalık aktivitesi KRK’nın sıklığı ile korele olmadığı gösterilmiştir (63, 64). Ancak endoskopik ve/veya histolojik ölçülere dayanan aktif inflamasyon derecesini ölçmenin daha doğru olduğu bildirilmiştir. Bir İBH merkezinde yapılan bir retrospektif vaka kontrol çalışması KRK sıklığı ile histolojik analizle tanımlanan aktif inflamasyonun daha yüksek derecesi ile ilişkilendirilmiştir (65). Bu çalışmada kanser veya displazili hastalar aynı cinsiyet ve hastalığın benzer süresi ve yaygınlığı olan 2 displazisiz kişi (kontrol) ile karşılaştırılmıştır. Histolojik analize dayalı inflamasyonun şiddeti kolorektal neoplazi sıklığı için bir önemli değişken iken, bir multivariable modelde ise diğer değişkenler kontrol edildikten sonra; 5 puanla derecelendirilen inflamasyonun her bir ünitesi displazi ve kanser için riski 4,7 kat arttığı bulunmuştur. Ancak univaryant analizde anlamlı olan endoskopi ile ölçülen inflamasyon multivaryable modelde kanser riskinin anlamlı bir belirleyicisi olmadığı bildirilmiştir. Önemli olarak kanserli olgularla sınırlandırılan analiz (bu yüzden displazili olgular analizden çıkartılmıştır) histolojik inflamasyon ile kanser arasındaki ilişkiyi teyit etmiştir. Bu vaka kontrol çalışması histolojik inflamasyon için yeni bir skorlama bildirmesi ve diğerleri zaman ile inflamasyon durumunu (ortalama skor kullanmışlardı) hesaba katmamalarına rağmen histoloji ile inflamasyonun derecesi KRK için bir risk faktörü olarak kabul edilmiştir. Bu bulgular inflamasyon ölçümü için iyi kurgulanmış sistem kullanan ve yalnızca surveyans altında olan ve displazisiz olduğu saptanan hastaları içeren bir retrospektif çalışma ile teyit edilmiştir (66). Bu çalışmada histolojik inflamasyonu sınıflamak için 4 farklı evreleme sistemi kullanılmıştır. Buna göre histolojik aktivite indeksi evreleri şunlardır; i. İnaktif Kolit: Kriptit veya kript absesi yok. ii. Hafif Aktif Kolit: Örneklenmiş kriptlerin veya kesitlerin %50’sinden azında nötrofil infiltrasyonu veya ülserler veya erozyonlar yok. iii. Orta Aktif Kolit: Örneklenmiş kriptlerin veya kesitlerin %50’si ve fazlasında nötrofil infiltrasyonu veya ülser veya erozyon yok. iv. Ciddi Aktif Kolit: Diğer özelliklere bakılmaksızın ülser veya erozyonların olmasıdır. 10 İnflamasyon skorunda her bir birim artışı zaman içinde yüksek derece displazi veya KRK için riskte 3.8 kat artışa neden olduğunu gösterilmiştir (66). KRK aşikar kolitli bölgelerin proksimalindeki mikroskobik kolitli bölgelerde oluşabilmesi kanser riskinin saptanmasında kolonoskopiden daha ziyade histolojik inflamasyon bulgusunun daha önemli olduğu düşüncesini destekler (67). Çok ciddi inflamasyonlu hastalar tıbbi tedaviye yanıt vermedikleri için hastalığın erken evresinde sıklıkla kolektomiye gider. Böyle hastalar artık kolorektal kanser için riskte olmadıkları için bu ciddi kolitin artmış KRK ile ilişkili olmadığı yanlış izlenimini verebilmektedir. Erken evre kolektomiden kaçınmak için aktif inflamasyonun aşağıya yerleştiği hastalar KRK gelişmesi için risk altındadırlar. Histolojik olarak ölçülen aktif inflamasyon KRK için bir risk faktörü olduğu kanıtına rağmen, KRK gelişen İBH’lı çoğu hasta sessiz kronik inflamasyona sahiptir (60). 2.5. Patogenez Kolit ile ilişkili kanser kronik olarak inflame mukozadan gelişir ve displazi yok– belirsiz displazi–düşük dereceli displazi–yüksek dereceli displazi–karsinoma silsilesinde geliştiğine inanılmaktadır. Hastalarda tümör progresyonu bu basamakların bir veya daha çoğunu atlayabilir (61). Sporadik KRK gibi, İBH’lı hastada kolon karsinogenezi somatik hücrelerdeki mutasyonları takiben onların klonal genişlemesi gibi olaylar silsilesi yoluyla gerçekleşir. Ancak, kolonun 1 veya 2 odağında displaziden gelişen sporadik KRK’dan farklı kolitik mukozadan doğan kanser sıklıkla alan değişikliği etkisini gösteren multifokal displaziden gelişir. Genomik instabilitenin bir belirteci olan anöploid hücreleri takip etmek için, tekrarlayan kolonoskopik incelemenin kullanıldığı kronik ÜK’li hastaların çalışmaları hücre popülasyonlarının kolonun aynı konumunda kaldığını fakat zaman içinde yayılımın mukozanın daha geniş alanlarını işgal ettiğini gösterilmiştir (68). Üstelik, anöploidinin spesifik bir bölge içinde, anöploid hücrelerinin subklonları onların öncüllerinden ortaya çıktığı görülmektedir. Subklonlar, normal epitelin pahasına genişlediğinden giderek daha fazla miktarda bozulur. İBH’lı hastalarda çoğu kısım için neoplastik lezyonlar kolonik mukozanın inflame bölgeleri içinden doğar. Anormal klonlarla kolonik mukozanın geniş yamalarının gözlemlenen reepitelizasyonu kronik inflamasyondan ülserasyona iyileşme cevabının bir sonucudur. 11 Anöploidi displaziden sıklıkla daha yaygın olduğu için substansiyel genomik değişiklikler bozulmuş morfoloji olmaksızın kolonik mukozada ortaya çıkar (60). Kolit ilişkili kolon kanseri Anöploidi/CIN, MSİ Metilasyon COX-2 p53 mut. Normal mukoza p53 LOH DCC-DCP4 Erken adenom Ara adenom APC k-ras Geç adenom Kanser Şekil 2.1. Kolit ilişkili kolorektal kanserin moleküler patogenezi. APC: Adenomatöz polipozis koli, CIN: Servikal intraepitelyal neoplazi, COX: Siklooksijenaz, DCC-DCP4: Kolon ve pankreas kanserinde delesyon olan genler, DKK: Dickkopf protein ailesi, k-ras: Kirsten ras onkogen, LOH: Heterozigozite kaybı, MSI: Mikrosatellit instabilite (60). Sporadik kolorektal karsinom gelişimi ile ilişkili hücresel bozukluklar birçok kolit ile ilişkili kolon karsinogenez ile de ilişkilidir (69). Anöploidi/kromozomal instabilite, mikrosatellit instabilite (MSI), DNA metilasyonu, onkogen k-ras’ın aktivasyonu, COX-2 aktivasyonu, mutasyon ve sonuç olarak p53, APC ve DCC/DPC4 heterozigositesinin kaybını içeren yolaklar arasında benzerlik vardır. Ancak, kanserler arasında bu olayların sıklığı ve silsilesi farklıdır (69) (Şekil 2.1). Kolitle ilişkili kanserde kromozom instabilitesi ve MSI’nin sıklığı sırasıyla %85 ve %15’tir ve sporadik kolorektal karsinomadakine yaklaşık olarak benzerdir (70). Fakat sporadik kolorektal kanserin progresyonu bu değişikliklerin zamanlamasında ve sıklığında İBH ilişkili KRK’dan farklıdır (Şekil 2.2). Örneğin sporadik kolorektal karsinomanın progresyonunda ilk olay olan APC işlevinin kaybı, daha az sıklıktadır ve genellikle kolit 12 ilişkili displazi ve karsinomanın gelişmesinde geç ortaya çıkar (27,31,71). Displazi için negatif veya belirsiz olan kolitik mukoza hücrelerinde APC’deki mutasyonlar nadirdir, düşük dereceli displazili veya kanser dokularının %14’den azında APC’de mutasyonlara sahiptir (31,71,72). Ayrıca APC’nin allelik kaybı kolitle ilişkili neoplazmların %33’ünden daha azında görülmektedir (72). Sporadik kolon kanseri Anöploidi Metilasyon MSİ k-ras COX-2 APC Normal mukoza Erken adenom DCCDCP4 Ara adenom p53 Geç adenom Kanser Şekil 2.2. Sporadik kolon kanserinin moleküler patogenezi. APC: Adenomatöz polipozis koli, CIN: Servikal intraepitelyal neoplazi, COX: Siklooksijenaz, DCC-DCP4: Kolon ve pankreas kanserinde delesyon olan genler, DKK: Dickkopf protein ailesi, k-ras: Kirsten ras onkogen, LOH: Heterozigozite kaybı, MSI: Mikrosatellit instabilite (60). p53 işlevinin kaybı kolitle ilişkili kanserin progresyonunda önemli bir basamaktır. p53’ün allelik delesyonu kolitle ilişkili tümörlerin yaklaşık %50-%85’inde gözlemlenmektedir (73,74). p53’te heterozigositenin kaybı malign progresyonuyla ilişkilidir; displazisiz biyopsi örneklerinin %6’sında, belirsiz displazinin %9’unda, düşük dereceli displazinin %33’ünde, yüksek dereceli displazinin %63’ünde ve kanserlilerin %85’inde saptanmıştır (73). p53 mutasyonları kolitik kolon doku mukozasında bulunmuştur ve nondisplastik veya displazi için belirsiz mukozada sıktır (73,75). Dikkatli şekilde haritalanmış kolektomi örneklerinde p53 mutasyonu 13 anöploididen önce tümorogeneziste erken ortaya çıktığı saptanmıştır (75). Gerçekte p53 mutasyonları kronik inflamasyonun bu mutasyonlara sahip olabileceğini gösteren kanser olmayan ÜK hastalarının %50‘den fazlasından inflame mukozasında bulunmuştur (76). DNA metilasyonu kolitle ilişkili kanserin gelişimi ve progresyonuna da katkıda bulunur. Birkaç gendeki CpG adalarının metilasyonu displaziden öncedir ve ÜK’li hastaların mukozası boyunca saptanabilmektedir (13). Kolitli hastalardan neoplastik örnekler arasında hMLH1 hipermetilasyonun MSI’nin yüksek düzeyleri ile (%46) 6/13’ünde, MSI’nın düşük düzeyleri ile (%16) 1/6’sında ve MSI’sız (%15) 4/27’sinde gözlemlenmiştir, bu yüzden hMLH1 hipermetilasyonu MSI’a neden olabilir veya gelişimine katkıda bulunabilir (11). Hücre siklus inhibitörü p16INK4a’nın ekspresyonunun kaybı sporadik KRK ile ilişkilidir; bu gen ÜK hastalarının neoplastik örneklerinde sıklıkla hipermetillenmiştir (12). Displazi içermeyen biyopsi örneklerinin yaklaşık %10’u p16 promotorunun hipermetilasyonuna sahiptir; oran displazinin daha yüksek dereceleriyle artar, tümör örneklerinde %100’e ulaşır. p14ARF p53’ün indirekt regülatörüdür; p16INK4a gibi aynı gen tarafından kodlanır. Kodlayan genin hipermetilasyonu yoluyla, p14ARF ekspresyonunun kaybı adenokarsinomanın %50’sinde, displastiklerin %33’ünde ve ÜK’li hastalardan alınan displazisiz mukoza örneklerinin %60’ında bildirilmiştir (77). Genetik olarak kolon kanserine yatkın farede inflamasyonun oluşturulması neoplastik fenotipi uyarır. Örneğin, MSH2-/- farede dış tetikleyici olmaksızın spontan olarak kolon kanseri gelişmez. Ancak, dekstran sodyum sülfat (DSS) oral uygulaması ile inflamasyon oluşturulduğunda, 30 MSH2-/- farenin 28’i displazi veya kanser geliştirir, yüksek derecede displazi sıklığı MSH2-/-, MSH2+/- ve MSH2+/+ için sırasıyla %47, %23 ve %8 olduğu görülmüştür (78). MSH2-/- farede displastik lezyonlar dışında inflame ve inflame olmayan kolonik mukozanın MSI’ye de sahip olduğu, MSI kolonik inflamasyon yoluyla oluşturulabileceği ve insanlarda gösterildiği gibi displaziden önce meydana gelebileceği gösterilmiştir. DSS ile indüklenmeyen kolitli APC Min/+ farelerde kolon tümörleri gelişmemiştir (79), fakat bu farelere 1 defa DSS verildiğinde %22’sinde, 2 defa DSS verildiğinde %40’ında tümör gelişmiştir. Ayrıca, DSS uygulaması akut ve kronik inflamasyon bölgeleri yanı sıra iyileşmiş mukoza bölgelerinde displazinin yüksek sıklığıyla sonuçlanmıştır. Yüksek inflamasyon 14 skorlu hayvanlar kolitin APC Min/+ farede neoplazinin gelişimini hızlandırdığını gösteren anlamlı daha fazla displastik lezyonalara sahip olduğu bildirilmiştir (79). Kolon kanserinin kesin hücresel kaynağı hakkında bilgi azdır. Mutasyonlar prolifere olmayan farklılaşmış hücre tipinde ortaya çıkarsa mutasyonlar yayılmayacaktır. Kript tabanındaki transforme olmuş kök hücreler bu nedenle erken neoplastik lezyonların gelişimi ve devamından sorumlu olabileceği düşünülmektedir. Kolonik kök hücreden spesifik olarak APC’nin kaybı progresif olarak büyüyen neoplazmlarla sonuçlanır, oysaki (kript tabanına yakın yerleşmiş) transit amplifiye hücrelerden kayıp neoplazm ile sonuçlanmaz (80). APC Min/+ farede sulindak apopitozu uyararak kök hücreleri ortadan kaldırır (81). Epitel kök hücre bölümü yerel mikro çevreden önemli sinyaller alır ve kript tabanındaki epitel mezenkim etkileşimini bu bölüm sürdürür (82). Örneğin epimorfin kolon kök hücrelerinde etkisi olan myofibroblastlar ve intestinal mezenkimal hücrelerden yapılan bir antiproliferatif, promorfojenik faktördür. İnflamasyonu uyarmak için DSS verilen ve karsinojen azoksimetana (AOM/DSS) maruz kalan epimorfin yoksun fare vahşi tip fare ile karşılaştırıldığında kolon tümörü oluşumunun sıklığı ve yaygınlığı azaldığı görülmüştür (83). Kolitin uyardığı kolon kanserinde kök hücrelerin rolü hakkında bilgiler azdır, fakat kök hücrelerin kesin belirteçleri ile genetik çalışmalar daha fazla bilgi sağlayacaktır. Transkripsiyonel sessizleşme birçok organizma için genel fizyolojik bir süreçtir ve uygun gelişim için sıklıkla gereklidir (84). İnsan hücrelerinde, genellikle sessizleşme etkilenen genlerin promotorlerinde CpG zengin dizilerinin (CpG adaları) metilasyonunu içerir ve imprint ve X kromozomu inaktivasyonu için önemlidir. Metilasyon, sırayla; metillenmiş DNA bağlayıcı protein ve histon deasetilazları içeren bir protein kompleksi alımı ile ve en sonunda, transkripsiyon faktörlerinin dışlanması ile kapalı bir kromatin yapısının oluşumu ile sonuçlanır (85). Bu sessizleşme klonaldir ve somatik hücrelerde fizyolojik olarak geri dönüşümsüz olduğu düşünülmektedir. Neoplastik hücreler; hücre döngüsü kontrolü, DNA onarımı ve anjiyogenez gibi kritik süreçleri düzenleyen genleri içeren çeşitli genlerin susturulması ve sıklıkla anormal metilasyon gösterir (86). Neoplastik hücrelerde anormal promotor metilasyonunun nedeni (leri) tam olarak aydınlatılmış değildir. Wnt proteinleri hücre büyüme ve farklılaşmasını düzenleyen glikozillenmiş ve lipidlenmiş 19 proteinli bir ailedir (87,88). Wnt’ler embriyonun GPS sistemidir: vücut 15 aksını belirler ve organları doğru yönde büyümeye götürür. Yetişkin memelilerde Wnt’lerin kontrolü kemik iliği, barsak ve deri gibi kendini sürekli yenileyen dokuların idamesini kontrol eder (89,90). Bozulmuş Wnt sinyali gelişme bozuklukları ile sonuçlanır ve kolorektal kanser, osteoporoz ve nörodejeneratif bozukluklar gibi farklı insan patolojilerinin altında yatar (89,90). Farklı hücre içi sinyal iletim yollarının aktivasyonu yoluyla onların etkilerini ortaya çıkarır. Wnt’lerin belirgin sinyal çıkışı alıcı hücreler üzerinde mevcut hücre yüzey reseptörlerinin repertuvarına bağlı görünüyor. Örneğin yedi transmembran reseptörlerinin (7TMs) Frizzled ailesinin bir üyesine Wnt5a ve düşük dansiteli lipoprotein reseptörü ile ilişkili protein ailesinin (LRP5/6) bir temsilcisine bağlanır, Wnt duyarlı genlerin transkripsiyonu düzenleyen T-hücre faktörü (TCF) ile uyum içinde hareket eden β-kateninin stabilizasyonunu uyarır (91,92). Ancak bu reseptöre tirozin kinaz ROR2’yi bağlarsa Wnt-5a bu β-katenin-bağımlı yolu inhibe edebilir (92,93). Böylece reseptör düzeyleri ve onların reseptörlerine Wnt ligandlarının nisbi afiniteleri sinyal çıkışını belirler (94-96). β-katenin sinyal hiperaktivasyonunun özellikle kolon kanseri olmak üzere çeşitli kanserlerin bir tetikleyicisi olduğu ortaya çıkarılmıştır (89). Oysaki azaltılmış sinyal bozulmuş kemik oluşumu ve nörodejeneratif hastalıkların altında yatar (97). Sonuç olarak farklı endikasyonlar için bu yolağın inhibitörlerinin veya aktivatörlerinin etkisi olduğu düşünülmektedir. 2.6. Wnt/β-katenin sinyal yolağı Wnt/β-katenin sinyali sitoplazmada β-katenin degradasyonu sayesinde düzenlenir. Latent hücrelerde β-kateninin sitoplazmik ve nükleer fazlalığı destrüksiyon kompleksi olarak adlandırılan büyük bir proteinin toplanmasının etkisiyle düşük düzeyde tutulmaktadır. Destrüksiyon kompleksi bir tümör baskılayıcı genin Axin1/Axin2 ve APC içeren birkaç yapı proteinini içerir. Buna ek olarak bu Ser/Thr kinaz glikojen sentaz kinaz 3 (GSK3α/ GSK3β) ve kazein kinaz Iα (CKIα) içerir (87,88). De novo-sentezlenen β-katenin birkaç N-terminal serin ve treonin rezidülerinde CKIα ve GSK3 yoluyla sırayla fosforillendiği destrüksiyon kompleksine katılır. Bu fosforilasyonlar proteozom tarafından ve ubikuitinasyon ve sonraki destrüksiyon için βkatenini işaret eder. β-kateninin eksikliğinde TCF ailesinin proteinleri Wnt spesifik gen transkripsiyonunu baskılar (Şekil 2.3) (87,88). 16 Wnt/β-katenin sinyali Frizzled ailesinin bir üyesine, LRP5 veya LRP6’a Wnt’lerin bağlanmasıyla başlatılır. Sonuçta oluşan üçlü kompleks destrüksiyon kompleksinin birkaç bileşenlenini membrana toplar. Destrüksiyon kompleksinin fonksiyonu inhibe edilir ve fosforillenmemiş β-katenin sitoplazmada birikir ve sonunda nükleusa transloke olur. Burada Wnt duyarlı gen transkripsiyonu represör TCF aktivatöre dönüştürme TCF proteinleri ile ilişkilidir (87,88). WNT yolağı etkin değil WNT Yolağı WNT yolağı etkin W DKK 1 Frizzled LRP5/6 GBP LKB1 Ubikinasyon aracılı yıkım Aksin GSK3 APC Dsh β- tcf P βΒkatenin Hedef genler Kanser olmayan hücre CBP Kanser hücresi Şekil 2.3. Kanser ve kanser olmayan hücrede Wnt yolağının şematik gösterimi (98). 2.7. Kanserde Wnt/β-katenin sinyali 1990’ların başında, Wnt/β katenin sinyaline olan ilgi β-kateninin tümör süpresör APC’e bağlanmasının bulunmasıyla giderek artmaktadır (99). O zamandan beri APC genini içeren kromozomal bölgedeki delesyonlar çok sayıda intestinal polip ve sonunda tümör oluşumu ile karakterize ailesel adenomatöz polipozis diye adlandırılan bir herediter hastalıkla ilişkilendirilmiştir. APC’de fonksiyon kaybedici mutasyonlar sadece 17 ailesel adenomatöz polipozisin büyük çoğunluğunda bulunmadığı ayrıca sporadik kolorektal karsinomlarda da bulunduğu görülmüştür. Bu mutasyonlar tipik olarak β katenin, Axin1 veya Axin 2’nin bağlanmasının yetersiz olduğu kısalmış proteinlerin oluşumuyla sonuçlanır (89,100). O zamandan beri destrüksiyon kompleksinin diğer çekirdek bileşenlerinde mutasyonlar çok çeşitli kanserlerde varolduğu bildirilmiştir. Kolon karsinomalarının küçük bir kısmında ve melonomalarda β-katenin geninde mutasyonlar gözlemlenmiştir (3,19). Bu mutasyonlar ya β-kateninin N terminalinin delesyonuna yol açan kısalmalardır ya da GSK3α/β veya CKIα tarafından hedeflenen serin, treonini etkileyen nokta mutasyonlarıdır. Bu mutant β-katenin proteinleri fosforilasyona dirençlidir ve böylece proteozomal degredasyondan kaçarlar. Sonuçta βkatenin etkilenmiş hücre içinde birikir. Stabilize edilmiş ve nükleusa lokalize olmuş βkatenin hemen hemen tüm kolon kanserlerinde bulunur ve onkogen c-myc ve hücre siklusu regülatörü siklin D1 gibi proliferatif Wnt düzenleyici genler tümör progresyonuna katkıda bulunur (89,100). Wnt sinyal yolağı, kolon karsinogenezisi ve kolon kök hücre kısmının çoğalma ve farklılaşmasının kontrolünde yer alır (15,16). Wnt2, Wnt5a, frizzled (Fz) reseptörleri ve downstream transkripsiyonel düzenleyici LEF1’i içeren Wnt yolağının diğer bileşenlerinin mekanizması bozulduğu için bu yolağı aktive eden mutasyonlar kolon kanserinin sporadik formlarında % 80 daha fazla bulunmuştur. Bu yolak inflamatuvar barsak hastalığı (İBH) ile ilişkilendirilmiş kolon kanserinde veya genel olarak İBH’da önemli bir rol oynayıp oynamadığı araştırılmaya devam etmektedir (17,18,101,102). Wnt yoluyla sinyal bir Wnt ligandı ile başlar, serpantin hücre yüzey Fz reseptörü ve LRP5/6 koreseptör ile etkileşen salgılanan büyüme faktörü ile sinyal kaskadı başlatılır (19). Disheveled (insan terminolojide Dsh veya dvl diye adlandırılır) ailesinin üyeleri glikojen sentaz kinaz-3β’nın inhibisyonuna yolaçan Fz (20), APC ve axin etkileşimi, β-kateninin fosforilasyonu ve yıkımını önler. β-katenin sonra birikir ve MYC, siklooksijenaz-2 (COX-2) ve siklin D1 (CCND1)’i içeren birçok hedef genlerin transkripsiyonu başlatan HMG-box transkripsiyon faktörlerinin ailesi lenfoid-arttırıcıfaktör/T-hücre faktör (LEF / TCF) üyelerini bağlayan çekirdeğine geçer (21-24). βkatenin aracılı Wnt sinyalizasyon kanonikal Wnt yolu olarak adlandırılır. Sinyalizasyon ekstraselüler olarak Wnt ligandları bağlayan çözünür Fz ilişkili proteinler (sFRPs, FzBs) 18 tarafından ve LRP yüzey molekülü bağlayan dickkopf (DKK) ailesi proteinleri tarafından bloke edilebilir (25,26). β-katenin stabilizasyonu ile sonuçlanan ya APC yada β-katenin kendisindeki mutasyonlar sporadik kolon kanserinde son derece yaygın iken bu tür mutasyonlar ülseratif koliti olan hastaların tümörlerinde nadir olarak ortaya çıkmaktadır (şekil 2.4) (27,31,72,103). 1-hydrozyanthraquinone ve methylazoxymethanol asetat yoluyla indüklenen ülseratif kolit ile ilişkili rat-kolon karsinogenezinde β-katenin mutasyonları görülebilmesine rağmen APC mutasyonları görülmez (104-106). Uzun süredir ülseratif kolitliler kolon kanseri için yüksek riske sahiptir ve bu hastalardan alınan örneklerde, Crohn hastalığı olan hastaların iltihaplı mukozalarının, ülseratif kolitli hastaların iltihaplı barsak mukozaları ile karşılaştırıldığı nükleotid dizi analizi 3 Wnt yolağı geni SARP1 (SFRP2), frizzled (FZD) ve disheveled (DVL)’nin ekspresyonunun artmış olduğu gösterilmiştir (107). Kanserde Wnt sinyalizasyonu Tümör Baskılayıcı Gen Onkogen WNT 5A Wnt 1 Fz SFRP Dsh APC GSK AKSİN β katenin Frat/GBP Lgs/BCL 9 TCF 1 Şekil 2.4. Kanserde Wnt sinyalizasyonunda ilişkisi (108). 19 Wnt antogonist genleri tümör supresör olarak işlev görür ve bazı insan malignitelerinin patogenezine katkıda bulunur. Son zamanlarda promotor CpG hipermetilasyonu ve sFRP-1, sFRP-2, sFRP-4, ve sFRP-5, Dkk-3, ve Wif-1 genleri gen susturmada bazı insan kanserlerinde tespit edilmiştir (109,110). 20 3. GEREÇ ve YÖNTEM 3.1. Hasta ve Kontrol Gruplarının Toplanması Bu Çalışmada WNT yolağı genlerinin promotor metilasyonunun ve ekspresyonunun İBH ilişkili karsinogenezisin seyri sırasında ortaya çıkıp çıkmadığını ve ayrıca İBH ilişkili neoplazininin seyri sırasında metilasyon olaylarının ve ekspresyonunun progresyonu karakterize edip edemeyeceğini araştırıldı. Mersin Üniversitesi Yerel Etik Kurulu’ndan çalışma için onay alındı. Çalışmaya katılan bireylerin hepsi için bilgilendirilmiş onam formu oluşturuldu ve çalışma hakkında bilgi sahibi olmaları sağlanarak onayları alındı. Mersin Üniversitesi, Tıp Fakültesi, Dâhili Tıp Bilimleri, İç Hastalıkları Anabilim Dalı’nın Gastroenteroloji Bilim Dalı tarafından takip edilen ve distal tip ÜK tanısı almış ve sürveyans kolonoskopisi yapılan 20 bireyden hasta grubunu oluşturuldu. Kontrol grubu ise kolon kanseri taraması yapılan herhangi bir yakınması ve endoskopik patolojisi olmayan 15 kişiden oluşturuldu. Surveyans kolonoskopisi yapılan ÜK’lı hastalarda çekumdan başlandı ve her 10 cm. de bir 4 kadrandan 2’şer biyopsi alınarak anal kanala dek biyopsi örnekleri alındı. Bu biyopsiler tarama amaçlı histopatolojik incelemeye tabi tutuldu. Distal tip ÜK’li hastalarda biyopsiler, rektum 20. santimetreden ve çekuma 10 cm uzaklıktan her bir bölgeden en az 5 adet olmak üzere standart biyopsi forsepsi ile alındı. Sürveyans kolonoskopisi yapılan hasta grubunda malignite şüphesi var ise veya displazi ile ilişkili lezyon-kitle (DALM) şüphesi olan lezyonlardan ayrıca biyopsi alındı. ÜK’in endoskopik aktivitesi Rahmilevich endoskopik aktivite indeksine göre derecelendirildi. Sürveyans için alınan örnekler aynı zamanda histopatolojik incelemeye tabi tutuldu. Böylelikle distal tip ÜK’li hastalarının hem normal görünümlü hem de inflame kolonik mukozasından biyopsiler alınarak ÜK hastalarında inflame ve inflame olmayan mukoza arasında çalışılan Wnt yolağı genlerinin metilasyonu ve ekspresyonları açısından bir fark olup olmadığı araştırıldı. Kontrol grubu hastalarında da sürveyans kolonoskopilerindeki gibi aynı şekilde biyopsiler; rektum 20. santimetreden ve çekuma 10 cm uzaklıktan biyopsiler alındı. Alınan biyopsi materyallerinden RNA ve DNA izolasyonu yapıldı. Manuel yöntemle izole edilen RNA’lar kullanılarak cDNA’ları elde edildi. 21 3.2. RNA İzolasyonu ve RT-PCR Endoskopik kolon biyopsi örneklerinden RNA izolasyonu manuel olarak Asit Guanidinyum-Fenol-Kloroform Yöntemi (AGPC) ile yapıldı (111). 3.2.1. Kullanılan Solüsyonlar a. Sodyum Sitrat (0,75 M): 15,78 g sodyum sitrat tartıldı ve DEPC-H2O ile 100 ml’ye tamamlanarak hazırlandı. Oda ısısnda 6 ay stabildir. b. N-Lauroyl Sarkozil (%10): 1 gr sarkozil tartıldı ve DEPC-H2O ile 10 ml’ye tamamlanarak hazırlandı. Oda ısısnda 3 ay stabildir. c. DEPC-H2O: 1000 ml de-iyonize su içerisine 1 ml DEPC eklendi. 1 gece oda ısısında bekletildi. Ertesi gün, 60oC’de 2 saat şişenin kapağı hafifçe gevşetilerek bekletildikten sonra otaklavlandı. +4 oC’de 1 yıl stabil kalabilir. d. Guanidinyum İsotiyosiyonat (GSCN) Solusyonu: 25 mg GSCN (4 M), 5 ml sodyum sitrat (0,75 M), 2,5 ml sarkozil (%10) tartıldı ve DEPC-H2O ile 50 ml’ye tamamlanarak hazırlandı. Oda ısısnda 3 ay stabildir. e. Denatürasyon Solüsyonu: 9,9 ml GSCN çözeltisi içerisine 0,1 ml 2merkaptoetanol (14 M) eklenerek hazırlandı. Oda ısısnda 1 ay stabildir. 3.2.2. Yöntem RNA izolasyonu için steril ependorftaki doku homojenize edilip üzerine 500 l denatürasyon solüsyonu eklenip birkaç saniye vortekslendikten sonra 15 dk -20oC’de bekletildi. 700 l fenol-kloroform-isoamilalkol (25:24:1) (UltraPure™ Phenol: Chloroform: Isoamyl Alcohol (25:24:1, v/v) 15593-031) eklenip, 15 dk -20oC’de bekletildi. 4 dk 10.000 rpm’de santrifüjlendi. Üst faz yeni bir 1,5 ml’lik tüpe alındı ve üzerine 700 l kloroform-isoamilalkol (24:1) (Ambresco x205 Chloroform: Isoamyl Alcohol 24:1) eklendi. 2 dk 10.000 rpm’de santrifüjlendi. Üst faz yeni bir 1,5 ml’lik ependorf tüpe alınarak üzerine 1 ml absolu etanol eklendi ve en az 1 gece bekletilmek üzere -20oC’ye bırakıldı. 15 dk 12.500 rpm’de santrifüjlendi. Tüpteki etanol dökülüp, 500 l %70’lik etanol eklendi ve birkaç saniye vortekslendi. 5 dk 12.500 rpm’de 22 santrifüjlendi. Süpernatant atılıp, tüpün ağzı açık olarak en az 10 dk bekletildi. Pelet üzerine 50 l DEPC-H2O eklendi. RNA’nın çözülmesi için 70oC’de 1 saat bırakıldı. Elde edilen total RNA, 200 ünite Moloney Murine Leukemia Virus Reverse Transkriptaz (MMLV-RT) enzimi kullanılarak cDNA’ya reverse transkribe edildi. Bunun için elde edilen RNA örnekleri ile (1 µl) RT-PCR yapıldı. PCR ortamı; 5 µl 5X RT-PCR buffer,(Fermentas) 10 µl dNTP (2 mM Fermentas) karışımı, 1µl poli-T primeri (05μg/μl Metabion), 0,1 µl reverse transkriptaz enzimi (200u/µl Fermentas), 0,5 µl RNAse inhibitörü (40u/µl Fermentas) ve 12 µl distile su ile hazırlandı. RT-PCR koşulları; 37 oC’de 1 saat 95 oC’de 5 dakika Elde edilen cDNA’lar analizi yapılana kadar +4 oC’de saklandı. 3.2.3. Real-Time PCR-Comparative CT (ΔΔCt) Metodu ile Ekspresyon Analizleri Biyopsi örneklerinin RNA’larından cDNA eldesi yapılarak, SFRP2, SFRP4, SFRP5, APC1, APC2 ve house keeping gen olan beta aktin’in (ACTB) ekspresyonları Real-Time PCR (ABI 7500, Applied Biosystems) cihazı ile ve “Comparative CT (ΔΔCT)” analizi yapılarak belirlendi (112-114). Bu çalışma hazırlığı için bu genlerin her birinin primer ve prob dizaynı yapılıp sentezletildi. 3.2.4. Kullanılan Primer ve Problar Primer ve problar, “Metabion International AG, D-82152 Martinsried/Deutschland” tarafından sentezlendi. Kullanılan primer ve problar çizelge 3.1. de gösterilmiştir. 23 Çizelge 3.1. Real-Time PCR için kullanılan primer ve problar. Gen Adı Diziler SFRP2 Forward 5’- TGCTTGAGTGCGACCGTTT -3’ NCBI Reference Sequence: NM_003013.2 SFRP4 Reverse 5’- CAGGAGGTGGTCGCTGCTA -3’ NCBI Reference Sequence: NM_003014.3 SFRP5 Reverse 5’- TGATAGGGTCGTGCAGGAACT -3’ NCBI Reference Sequence: NM_003015.3 Reverse 5’-TGGTGTCCTTGCGCTTCAG-3’ Probe 5’-Fam- ACAACGACCTTTGCATCCCCCTCG -BHQ-1-3’ Forward 5’- GCCGTGCTGCGCTTCTT -3’ Probe 5’-Fam- CCATGTACGCGCCCATTTGCAC -BHQ-1-3’ Forward 5’- TGATTGGAGCCCAGAAAAAGA -3’ Probe5’-Fam-AAGCTGCTCAAGCCGGGCCC-BHQ-1-3’ APC1 Forward5’-GAACCTGGAGGTACTTCATGTGAA-3’ NCBI Reference Sequence: NM_022662.2 APC2 Reverse5’-GCAAATGTTTGGCCCACTAAA-3’ Probe5’-Fam-AGCGCTTCGTTCAAACAGCAATCCA-BHQ-1-3’ Forward5’-CATCAGCGAGCGGATGTG-3’ NCBI Reference Sequence: NT_011255.14 ACTB Reverse5’-GCAGTTCTCGATGCAGATGGA-3’ NCBI Reference Sequence NM_001101.3 Reverse5’-GCCGATCCACACGGAGTACT-3’ Probe5’-Fam-CCTCGAACTCAAGCTGCTGCCGG-BHQ-1-3’ Forward5’-GGCACCCAGCACAATGAAG-3’ Probe5’-YakimaYellow-TCAAGATCATTGCTCCTCCTGAGCGC-BHQ-1-3’ 3.2.5. Real-Time PCR Reaksiyon Ortamının Hazırlanışı Real-Time PCR reaksiyon ortamını, total 25 µl olacak şekilde hazırlandı: a. 12.5 µl, 2X TaqMan Gene Expression Master Mix (Applied Biosystems) 24 b. Son konsantrasyon; SFRP2R, SFRP2F, SFRP4R, SFRP4F, SFRP5R, SFRP5F, APC1R, APC1F, APC2R, APC2F, ACTB-F ve ACTB-R primerlerinden 900 nmol olacak şekilde primerler kullanıldı. FAM ile işaretli SFRP2-Pr, SFRP4-Pr, SFRP5-Pr, APC1-Pr ve APC2-Pr problarından ve Yakima Yellow ile işaretli ACTB-Pr probundan 200 nmol olacak şekilde problar kullanıldı. c. 2,5 µl cDNA örneği d. 5 µl Distile Su 3.2.6. Real-Time PCR Reaksiyon Şartları 50ºC’de 2 dakika ön inkübasyon (1 döngü) 95ºC’de 10 dakika aktivasyon (1 döngü) 95ºC’de 15 saniye denatürasyon (50 döngü) 60ºC’de 1,5 dakika bağlanma/uzama (50 döngü) Real Time PCR işlemi ve kantitasyon “ABI Prism 7500 Real-Time PCR System (Applied Biosystems)” cihazı ve “7500 Software; SDS 2.0.3 software for 7500 Real Time PCR Product (Applied Biosystems)” programı kullanılarak gerçekleştirildi. 3.2.7. Ekspresyon Tayini Primer ve problar kullanılarak Gerçek Zamanlı Polimeraz Zincir Reaksiyonu (Real Time Polymerase Chain Reaction) yöntemiyle “Comparative CT (ΔΔCT)” analizi yapılarak SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin kantitasyonu belirlendi. Kontrol geni olarak insan ACTB kullanıldı. Comparative CT analiziyle ekspresyon tayini yapabilmek için ayrıca bir internal kontrol olarak standart bir RNA’dan (Taqman Control-Human RNA, 50ng/µl) hazırlanan cDNA kullanıldı. 25 3.3. DNA İzolasyonu Çalışmamızda hasta ve kontrol grubuna ait bireylerden alınan endoskopik kolon biyopsi örnekleri 1,5ml’lik steril santrifüj tüplerine her bireye ait özel protokol numaraları kaydedilerek -80oC’de saklandı. DNA izolasyonu için High Pure PCR Template Preparation Kit (Roche) kullanılarak aşağıda sıralanan yöntem uygulandı: 1. Steril santrifüj tüpü içerisine konan endoskopik kolon biyopsi örnekleri homojenize edilip üzerine 200 l Doku Parçalama Solüsyonu ve 40l Proteinaz K Solüsyonu eklendi, dikkatlice vortekslenerek karışması sağlandı. 2. Örnekler 55 oC’de yarım saat inkübasyona bırakıldı. 3. İnkübasyonu tamamlanan örneklerin üzerine 200 l bağlanma (binding buffer) solüsyonu eklendi ve vortekslenerek karıştırıldı. 10 dakika 70 o C’de inkübasyona bırakıldı. 4. İnkübasyondan alınan karışımın üzerine 100 l izopropanol eklendi ve vortekslenerek karıştırıldı. 5. Hazırlanan karışım 2 ml’lik toplama tüpüne yerleştirilmiş kolona aktarıldı ve 8000 rpm’de 1 dk santrifüj edildi, toplama tüpü atılarak kolon yeni toplama tüpüne yerleştirildi. 6. Kolon üzerine 500 l Wash Buffer I pipetlendi ve 8000 rpm’de 1dk santrifüj edildi, toplama tüpündeki sıvı boşaltılarak kolon tekrar yerleştirildi. 7. Kolon üzerine 500 l Wash Buffer II pipetlendi ve maksimum hızda (>12000 rpm) 3 dk santrifüj edildi, toplama tüpü atılarak kolon 1,5 ml’lik steril saklama tüpüne yerleştirildi. 8. Kolon üzerine 200 l Elution Buffer pipetlendi. 2 dk oda ısısında bekletildikten sonra 8000 rpm’de 1dk santrifüj edildi, Kolon atıldı ve elde edilen DNA +4 oC’de saklandı. 26 3.3.1. DNA Modifikasyonu Elde edilen DNA’lar EZ DNA Metilasyon Gold Kit (Zymo Research) kullanılarak üretici firmanın önerdiği yöntemle metilasyon dönüşümleri gerçekleştirildi. Bu işlem için aşağıdaki adımlar takip edildi; 1. PCR tüpünün içine 130 μl CT conversion reagent ve 20 μl DNA örneği konur ve örnek tüpleri thermocycler’da; 98 oC’de 10 dakika 64 oC’de 2,5 saat PCR yapıldı. 2. Zymo-Spin kolonlarına 600 μl M- Binding buffer konur ve spinependorf tüpünün içersine yerleştirilir. 3. Thermocycler içindeki örnekler M- Binding buffer içeren Zymo-Spin kolon içersine kondu ve birkaç kez ters-düz etmek suretiyle karıştırıldı. 4. 10000 rpm’de 1 dk santrifüj edildi ve alttaki sıvı uzaklaştırıldı. 5. Kolon içine 100 μl M-Wash buffer eklendi 12500 rpm’de 45 saniye santrifüj edildi. 6. Kolon içine 200 μl M-desulphination buffer ilave edildi oda sıcaklığında 15 dakika inkübe edildikten sonra 10000 rpm'45 saniye santrifüj edildi. 7. Kolon içine 200 μl M-Wash buffer konulup ve 10000 rpm’de 30 saniye santrifüj edildi. 8. Tekrar 200 μl M-Wash buffer eklendi ve 10000 rpm’de 45 saniye santrifüj edildi. 9. Kolon 1,5 ml’lik mikrosantrifüj tüpüne yerleştirildi 10 μlM- Elution buffer kolonun matriksine eklenip 13500 rpm’de 1 dk santrifüj edildi. Elde edilen modifiye DNA’lar -20 o C’de saklandı. WNT yolağı genlerinin metilasyon durumunu MSP (Metilasyon Spesifik Polimeraz Zincir Reaksiyonu) yöntemi ile araştırıldı. MSP; Sitozinlerdeki metilasyon durumunun tespiti için geliştirilen metotlardan biridir. 27 3.3.2. Metilasyon Spesifik PCR İçin Kullanılan Primerler Bu çalışma kapsamında öngörülen genlerin metilasyon profilinin MSP yöntemi ile tespit edilmesi için kullanılan primer dizileri Çizelge 3.2.’de verilmektedir. Çizelge 3.2. Metilasyon Spesifik PCR İçin kullanılan primerler Metile F 5’-GGGTCGGAGTTTTTCGGAGTTGCGC-3’ R 5’-CCGCTCTCTTCGCTAAATACGACTCG -3’ SFRP2 Unmetile F 5’- TTTTGGGTTGGAGTTTTTTGGAGTTGTGT -3’ R 5’- AACCCACTCTCTTCACTAAATACAACTCA-3’ Metile F 5’- GGGTGATGTTATCGTTTTTGTATCGAC-3’ R 5’- CCTCCCCTAACGTAAACTCGAAACG-3’ SFRP4 Unmetile F 5’- GGGGGTGATGTTATTGTTTTTGTATTGAT-3’ R 5’- CACCTCCCCTAACATAAACTCAAAACA-3’ Metile F 5’- AAGATTTGGCGTTGGGCGGGACGTTC-3’ R 5’- ACTCCAACCCGAACCTCGCCGTACG-3’ SFRP5 Unmetile F 5’- GTAAGATTTGGTGTTGGGTGGGATGTTT-3’ R 5’- AAAACTCCAACCCAAACCTCACCATACA-3’ Metile F 5’- TATTGCGGAGTGCGGGTC-3’ R 5’- TCGACGAACTCCCGACGA-3’ APC1 Unmetile F 5’- GTGTTTTATTGTGGAGTGTGGGTT-3’ R 5’- CCAATCAACAAACTCCCAACAA-3’ Metile F 5’- GTCGTTTGTTTAGGTTCGGATC-3’ R 5’- GACCCGAAATAACCTCGAAACG-3’ APC2 Unmetile F 5’- TGGTAGTGTTGTTTGTTTAGGTTTGGATTG-3’ R 5’- ACCAAAAATCCCAACCCAAAATAACCTCAAAACA-3’ 28 3.3.3. Metilasyon Spesifik PCR Ortamının Hazırlanışı Metilasyon profillerinin tespiti için içeriğinde Hot-Start Taq Polimeraz (5 U/μl ZymoTaq™ DNA Polymerase, E2002), 2X Reaksyon buffer (ZymoTaq™ E2002), dNTP (25mM ZymoTaq™ E2002), 25 mM MgCl2 (Fermentas), her bir gen için Primer M (Metabion International AG) ve Primer U (Metabion International AG), Bisulfid DNA1μl olacak şekilde 25 μL reaksiyon karışımı hazırlandı. Pozitif ve negatif kontroller için M kontrol DNA olarak (Chemicon International 90394) ve U kontrol DNA olarak (Chemicon International 90393) kullanıldı. 3.3.4. Metilasyon Spesifik PCR Şartları 95ºC’de 5 dakika 95ºC’de 30 saniye Annealing 30 saniye 72ºC’de 30 saniye 72ºC’de 5 dakika Denatürasyon 35 siklus Amplifikasyon Son uzama 100 ml 1XTBE (Tris-Borat-EDTA) tamponu içerisinde 2 gr agaroz plus mikrodalga fırında eritildikten sonra 0.5 g/ml konsantrasyonunda olacak şekilde etidyum bromid eklenerek, %2’lik agaroz hazırlandı. Elde edilen MSP ürünleri standart U ve M kontrol eşliğinde, %2’lik agaroz jelde 120 Volt elektrik akım uygulanarak elektroforez işlemi yapıldı. Jel Görüntüleme Sistemi (Vilber Lourmat Marne La Vallée, France) ile sonuçlar değerlendirildi. 3.3.5. Metilasyon Spesifik PCR Annealing Dereceleri ve Ürün Boyutları Çalışılan SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin metile ve unmetile primerlerle ile yapılan MSP’nin annealing dereceleri ve ürün boyutları Çizelge 3.3’de gösterilmiştir. 29 Çizelge 3.3. Metilasyon Spesifik PCR Annealing Dereceleri ve Ürün Boyutları Gen Adı Annealing Derecesi Ürün Boyutu SFRP2-M 58 ºC 138 bç SFRP2-U 64 ºC 145 bç SFRP4- M 64 ºC 111 bç SFRP4-U 64 ºC 115 bç SFRP5 -M 64 ºC 136 bç SFRP5-U 64 ºC 141 bç APC1-M 61 ºC 98 bç APC1-U 61 ºC 108 bç APC2-M 64 ºC 98 bç APC2-U 64 ºC 102 bç 30 3.4. İstatistiksel Analiz Çalışmanın istatistiksel analizleri Mersin Üniversitesi Tıp Fakültesi Biyoistatistik ve Tıbbi Bilişim Anabilim Dalı tarafından yapılmıştır. Yaş ve ekspresyon düzeyleri bakımından ülseratif kolit hastaları ile kontrol gruplarının karşılaştırmalarında ve hastalarda ekpresyon düzeylerinin metilasyon gruplarına göre yapılan karşılaştırmalarında Independent Samples t test kullanılmıştır. Kategorik değişkenlerin analizinde Ki-kare veya Likelihood ratio testinden yararlanılmıştır. İstatistik analizler SPSS v.11.5 paket programı ile yapılmıştır. İstatistik analizlerde p<0,05 ise sonuçlar anlamlı kabul edilmiştir. Sürekli değişkenler için tanımlayıcı istatistikler ortalama ve standart sapma cinsinden, kategorik değişkenler için ise frekans ve yüzde cinsinden tablo halinde verilmiştir. 31 4. BULGULAR Ortalama 13.65 yıldır distal tip ÜK tanısı almış 20 bireyden hasta grubu oluşturuldu. Kontrol grubu kolon kanseri taraması yapılan herhangi bir yakınması ve endoskopik patolojisi olmayan 15 bireyden oluşturuldu. İncelenen hasta ve kontrol grupları arasında yaş bakımından dağılımları; hasta grubunda 53,5±12,9 kontrol grubunda 53,6±9,8 idi ve bu fark istatistiksel olarak anlamlı değildi (p= 0.967). Değerlendirilen hasta grubundaki 20 bireyin 16’sı erkek (%80), 4’ü kadın (%20) ve kontrol grubundaki 15 bireyin 7’si erkek (%46,7), 8’i kadın (%53,3) idi. Bu hastaların endoskopik olarak 7’sinde splenik köşeden itibaren anüse dek submukozal damar ağı distorsiyone ya da submukozal damar ağı seçilmiyordu, Rahmilevich endoskopik aktivite indeksi 4 ve altındaydı. 12’sinde Rahmilevich endoskopik aktivite indeksi 6 idi ve 1’inde Rahmilevich endoskopik aktivite indeksi 8 idi. Rahmilevich endoskopik aktivite indeksi 8 olan hastada çıkan kolon proksimalinde (burada Rahmilevich endoskopik aktivite indeksi 1 idi) 1santimetrelik polip görüldü, çıkartılan polip materyalinin histopatolojik incelemesinde düşük dereceli displazi saptandı. Histopatolojik incelemede distal kolondan alınan biyopsi örneklerinde 13 hastada şiddetli aktif inflamasyon varken, 7 hastada ise hafif inflamasyon vardı. Proksimal kolonundan alınan biyopsi örneklerinde 12 hastada hafif aktif inflamasyon varken 8’inde inflamasyon yoktu. Proksimal kolonda inflamasyonu olmayan 8 hastanın 4’ünde distal kolonda hafif, diğer 4 hastada da şiddetli aktif inflamasyon vardı. (Çizelge 4.1). 4.1. Metilasyon Spesifik PCR Bulgularının Değerlendirilmesi Hastaların ve kontrollerin distal kolonundan alınan örneklerinin SFRP2, SFRP4, SFRP5, APC1ve APC2 genlerinin MSP ile değerlendirildiğinde metile primer ile bant görünen örnekler homozigotmetile, unmetile primer ile ile bant görünen örnekler unmetile ve hem metile primer hemde unmetile primer ile ile bant görünen örnekler ise heterozigot metile olarak kabul edildi. İstatistiksel değerlendirmede metile olanlar ve heterozigot metile olanlar metile olarak değerlendirildi. 32 4.1.1. SFRP2 Hastaların ve kontrollerin distal kolonundan alınan örneklerinin SFRP2 geninin MSP ile değerlendirildiğinde 20 hastadan 19’u metile (%95), 1 kişi unmetile (%5); kontrollerin ise 13’ü (%86,7) metile, 2’si unmetile (%13,3) idi (Şekil 4.1; Şekil 4.2). Hasta ve kontroller arasında distal kolonun metilasyonu açısından istatistiksel olarak anlamlı bir fark bulunmadı (p=0,385). Proksimal kolondan alınan örneklerin SFRP2 geninin MSP ile değerlendirilmesinde 20 hastadan 17’si metile (%85), 3’ü unmetile (%15) iken; 15 bireyden oluşan kontrol grubunun 14’ü metile (%93,3), 1’i unmetile (%6,7) idi. Proksimal kolondaki metilasyon oranları bakımından hasta ve kontrol grupları arasında istatistiksel olarak önemli bir farklılık saptanmadı (p=0,431). Hastaların distal ve proksimal kolonları kendi içinde değerlendirildiğinde ise distal kolonunda metilasyon olan 19 kişiden 16’sının (%84,2) proksimal kolonunda da metilasyon bulundu. Şekil 4.1 SFRP2 geninin metile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (138 bç), M: Markır (50bç). 1-4 ve 6-8 numaralı örneklerde metilasyon gözlenirken 5 numaralı örnekte metile primer ile metilasyon gözlenmedi. 33 Şekil 4.2 SFRP2 geninin unmetile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (145 bç), M: Markır (50bç). 1-4, 6-8 numaralı örnekler unmetile primer ile unmetile bant gözlenirken 5 numaralı örnek unmetile primer bant gözlenmedi. 4.1.2. SFRP4 Hastaların distal kolonundan alınan örneklerde 19 hasta metile (%95) ve 1 hasta unmetile (%5) iken; kontrollerin 14’ü (%93,3) metile, 1’i unmetile (%6,7) bulundu (Şekil 4.3; Şekil 4.4). Hasta ve kontrol gruplarında distal kolondaki metilasyon oranları arasında istatistiksel olarak anlamlı bir farklılık yoktu (p=0,834). Proksimal kolondan alınan örneklerde ise hastalardan 12’si metile (%60) ve 8’i unmetile (%40), kontrollerin 14’ü (%93,3) metile ve 1’i unmetile (%6,7) idi. Metilasyon oranları bakımından proksimal kolon örnekleri arasında istatistiksel olarak anlamlı bir fark bulundu (p=0,018). Hastaların proksimal kolon örneklerinde kontrollere göre metilasyon azalmıştı. Distal kolonu metile olan 19 kişinin 11’nin (%57,9) proksimal kolonunda da metilasyon bulundu. 34 Şekil 4.3. SFRP4 geninin metile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, 1-5,6,7 numaralı örnekler metile primer ile metile bant gözlenirken 5,8 numaralı örneklerde metile primer ile metile bant gözlenmedi. NK: Negatif kontrol, PK: Pozitif kontrol (111 bç), M: Markır (50bç). Şekil 4.4. SFRP4 geninin unmetile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (115 bç), M: Markır (50bç). 1,2,4,5,6 ve 7 nolu örnekler unmetile primer ile bant görülürken 3 numaralı örnekte unmetile bant gözlenmedi. 4.1.3. SFRP5 Hasta ve kontrollerin distal kolonlarından alınan örneklerde; 20 hastanın 12’si metile (%60) ve 8’i unmetile (%40), kontrollerin ise 7si (%46,7) metile, 6’sı unmetile (%53,3) idi (Şekil 4.5; Şekil 4.6). Hasta ve kontrol gruplarında distal kolondaki metilasyon oranları arasında istatistiksel olarak anlamlı bir farklılık yoktu (p=0,433). 35 Proksimal kolondan alınan örnekler incelendiğinde ise hastalarda 13 kişi metile (%65) ve 7 kişi unmetile (%35) bulundu. Kontrollerde ise 8 kişi metile (%53,3) ve 7 kişi unmetile (%46,7) idi. Proksimal kolondaki metilasyon oranları bakımından hasta ve kontrol grupları arasında istatistiksel olarak önemli bir farklılık saptanmadı (p=0,486). Hastaların distal kolonunda metilasyon olan 12 kişiden 8’inin (%66,7) proksimal kolonunda da metilasyon gözlemlendi. Distal ve proksimal kolonlardaki metilasyon oranları arasında istatistiksel olarak anlamlı bir fark gözlenmedi (p=0,848). Şekil 4.5. SFRP5 geninin metile primer ile PCR görüntüsü. 1-9 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (136 bç), M: Markır (50bç). 1-9 nolu örneklerde metile primer ile metile bant gözlendi. Şekil 4.6. SFRP5 geninin unmetile primer ile PCR görüntüsü. 1-9 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (141 bç), M: Markır (50bç). 1-9 nolu örnekler unmetile primer ile unmetile bant gözlemlenirken 10 numaralı örnekte unmetile bant gözlenmedi. 36 4.1.4. APC1 Distal kolondan alınan örneklerin incelemesi sonucu 20 hastanın 4’ü metile (%20), 16’sı unmetile (%80) iken kontrollerin 7’si metile (46,7) ve 8’si unmetile (%53,3) idi (Şekil 4.7; Şekil 4.8). Distal kolonda hasta ve kontroller arasında metilasyon açısından istatistiksel olarak anlamlı fark gözlenmedi (p=0,093). Proksimal kolondan alınan örnekler incelendiğinde ise 11 hasta metile (%55), 9 hasta unmetile (%45) ve kontrollerin 10’u metile (%66,7) ve 5’i unmetile (%33,3) bulundu. Hasta ve kontroller arasında metilasyon durumu açısından proksimal kolonda istatistiksel olarak anlamlı fark bulunmadı (p=0.486). Distal kolonu metile olan 4 hastadan 1’nin (%25) proksimal kolonunda da metilasyon gözlemlendi. Distal ve proksimal kolonlardaki metilasyon oranları arasında istatistiksel olarak anlamlı bir fark gözlenmedi (p=0.173). Şekil 4.7. APC1 geninin metile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (98 bç), M: Markır (50bç). 2, 6-9 numaralı örneklerde metile primer ile metile bant görülürken 3-5 numaralı örneklerde metile bant görülmedi. Şekil 4.8. APC1 geninin unmetile primer ile PCR görüntüsü. 1-7 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (108 bç), M: Markır (50bç). 1-3 ve 5-7 numaralı örneklerde unmetile primer ile unmetile bant görülürken 4 numaralı örnekte unmetile primer ile bant görülmedi. 37 4.1.5. APC2 Hastaların distal kolonundan alınan örnekler incelendiğinde 8 kişi metile (%40) ve 12 kişi unmetile (%60) iken kontrollerin 1’i metile (%6,7), 14’ü unmetile (%93,3) idi (Şekil 4.9; Şekil 4.10). Hasta ve kontroller arasında distal kolonda metilasyon bakımından istatistiksel olarak anlamlı fark bulundu (p=0,018). Hastaların distal kolonunda kontrollere göre metilasyon artmıştı. Proksimal kolonda ise hastaların 6’sı metile (%30), 14’ü unmetile (%70) iken kontrollerin 2’si metile (%13,3) ve 13’ü unmetile (%86,7) bulundu. Proksimal kolon örneklerinde metilasyon bakımından fark istatistiksel olarak anlamlı değildi (p=0,235). Distal kolonu metile olan 8 hastadan 3’nün proksimal kolonuda metile idi. Distal ve proksimal kolonlardaki metilasyon oranları arasında istatistiksel olarak anlamlı bir fark gözlenmedi (p=0,552). Şekil 4.9. APC2 geninin metile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (98 bç), M: Markır (50bç).3,4 ve 6-8 numaralı örnekler metile primer ile metile bant gözlenirken 1,2 ve 5 numaralı örnekler metile primer ile metile bant gözlenmedi. 38 Şekil 4.10. APC2 geninin unmetile primer ile PCR görüntüsü. 1-8 örneklerin görüntüsü, NK: Negatif kontrol, PK: Pozitif kontrol (102 bç), M: Markır (50bç). 1 ve 4-8 numaralı örnekler unmetile primer ile unmetile bant gözlemlenirken 2,3 numaralı örneklerde unmetile primer ile unmetile bant gözlemlenmedi. 4.2. Metilasyon ve İnflamasyon Bulgularının Değerlendirilmesi Hastaların distal ve proksimal kolonlarındaki inflamasyon bulgularını karşılaştırabilmek için; hastaların klinik ve inflamatuvar özellikleri, yaş, cinsiyet ve hastalık süreleri Çizelge 4.1’de verilmişitr. 39 Çizelge 4.1. Hastaların klinik ve inflamatuvar özellikleri. PMİ: Proksimal mikroskopik inflamasyon, DMİ: Distal mikroskopik inflamasyon, PEİ: Proksimal endoskopik inflamasyon, DEİ: Distal endoskopik inflamasyon, H: Hafif inflamasyon, Ş: Şidetli inflamasyon, E: Erkek, K: Kadın. Hasta PMİ DMİ PEİ DEİ Hastalık Süresi (yıl) Yaş Cinsiyet UK-1 YOK Ş YOK Ş 15 41 E UK-2 YOK Ş YOK H 18 46 E UK-3 H Ş YOK Ş 14 61 K UK-4 YOK Ş YOK Ş 12 36 K UK-5 H Ş YOK Ş 12 33 E UK-6 H H YOK H 15 59 E UK-7 H Ş YOK H 10 37 E UK-8 H H YOK Ş 15 47 E UK-9 YOK Ş YOK Ş 11 69 E UK-10 YOK H YOK H 15 56 E UK-11 H H YOK H 10 40 E UK-12 YOK H YOK Ş 13 49 E UK-13 YOK H YOK Ş 12 68 E UK-14 H Ş YOK Ş 16 62 E UK-15 H Ş YOK Ş 17 67 E UK-16 H Ş YOK Ş 13 80 E UK-17 H Ş YOK H 11 67 K UK-18 H Ş YOK H 10 53 E UK-19 H Ş YOK Ş 15 49 K UK-20 YOK H YOK Ş 19 50 E 40 Hastaların distal kolondan alınan örneklerin SFRP2 geninin metilasyon durumları ile mikroskobik inflamasyon durumu karşılaştırıldığında inflamasyonu olmayan grupta metile ve unmetile kişi gözlemlenmezken hafif inflamasyonu olan 7 kişi (%100) metile; şiddetli inflamasyonu olanlardan 12 kişi metile (%92,3) ve 1 kişi unmetile (%7,7) bulundu. (Şekil 4.11). Şekil 4.11. SFRP2 geninin distal kolon örneklerinin metilasyon durumları (metile, unmetile) ile distal mikroskobik inflamasyon durumlarının (yok, hafif, şiddetli) grafiksel gösterimi. Hastaların distal kolondan alınan örneklerin SFRP4 geninin metilasyon durumları ile mikroskobik inflamasyon durumu karşılaştırıldığında inflamasyonu olmayan grupta metile ve unmetile kişi gözlemlenmezken hafif inflamasyonu olan 7 kişi (%100) metile; şiddetli inflamasyonu olanlardan 12 kişi metile (%92,3) ve 1 kişi unmetile (%7,7) bulundu. (Şekil 4.12). 41 Şekil 4.12. SFRP4 geninin distal kolon örneklerinin metilasyon durumları (metile, unmetile) ile distal mikroskobik inflamasyon durumlarının (yok, hafif, şiddetli) grafiksel gösterimi. SFRP5 geninin distal kolon örneklerinde metilasyon ve mikroskobik inflamasyonu değerlendirildiğinde inflamasyonu olmayan grupta metile ve unmetile kişi gözlemlenmezken hafif inflamasyonu olan 4 kişi (%57,1) metile, 3’ü unmetile (%42,9); şiddetli inflamasyonu olanlardan 8 kişi metile (%61,5) ve 5 kişi unmetile (%38,5) bulundu. (Şekil 4.13). 42 Şekil 4.13. SFRP5 geninin distal kolon örneklerinin metilasyon (metile, unmetile) ile distal mikroskobik inflamasyon durumlarının (yok, hafif, şiddetli) grafiksel gösterimi. APC1 geninin distal kolon örneklerinde metilasyon ve mikroskobik inflamasyonu değerlendirildiğinde inflamasyonu olmayan grupta metile ve unmetile kişi gözlemlenmezken hafif inflamasyonu olan 3 kişi (%42,9) metile, 4’ü unmetile(%57,1); şiddetli inflamasyonu olanlardan 1 kişi metile (%7,7) ve 1 kişi unmetile (%92,3) bulundu. İstatistiksel olarak anlamlı değildi (p=0,065) (Şekil 4.14). 43 Şekil 4.14. APC1 geninin distal kolon örneklerinin metilasyon durumları (metile, unmetile) ile distal mikroskobik inflamasyon durumlarının (yok, hafif, şiddetli) grafiksel gösterimi. APC2 geninin distal kolon örneklerinde metilasyon ve mikroskobik inflamasyonu değerlendirildiğinde inflamasyonu olmayan grupta metile ve unmetile kişi gözlemlenmezken hafif inflamasyonu olan 2 kişi (%28,6) metile, 5’i unmetile(%71,4); şiddetli inflamasyonu olanlardan 6 kişi metile (%46,2) ve 7 kişi unmetile (%53,8) bulundu. İstatistiksel olarak anlamlı değildi (p=0,439) (Şekil 4.15). 44 Şekil 4.15. APC2 geninin distal kolon örneklerinin metilasyon durumları (metile, unmetile) ile inflamasyon (yok, hafif, şiddetli) durumlarının grafiksel gösterimi. 4.3. Real Time-PCR (Comparative CT) Analiziyle İlgili Bulgular Hasta ve kontrol grubunun proksimal ve distal kolonlarından alınan biyopsi örneklerinden elde edilen cDNA’lar ile Wnt yolağında yer alan SFRP2, SFRP4, SFRP5, APC1, APC2 ve ACTB’nin gen ekspresyonları Real-Time PCR (ABI 7500, Applied Biosystems) cihazı ile “Comparative CT (ΔΔCt)” analizi yapılarak belirlendi. Real-Time PCR cihazı ile her bir gen için elde edilen PCR amplifikasyon eğrileri ile her bir örneğin ekspresyon oranları değerlendirildi. (Sırasıyla ACTB, SFRP2, SFRP4, SFRP5, APC1 ve APC2; Şekil 4.16, Şekil 4.17, Şekil 4.18, Şekil 4.19, Şekil 4.20, Şekil 4.21). Elde edilen veriler SPSS (V.11.5) programına aktarılarak istatiksel analiz yapıldı. 45 Şekil 4.16. ACTB genine ait, YAKIMA YELLOW ile işaretlenmiş real time PCR amplifikasyon eğrisi. Şekil 4.17. SFRP2 genine ait, FAM ile işaretlenmiş real time PCR amplifikasyon eğrisi. 46 Şekil 4.18. SFRP4 genine ait, FAM ile işaretlenmiş real time PCR amplifikasyon eğrisi. Şekil 4.19. SFRP5 genine ait, FAM ile işaretlenmiş real time PCR amplifikasyon eğrisi. 47 Şekil 4.20. APC1 genine ait, FAM ile işaretlenmiş real time PCR amplifikasyon eğrisi. Şekil 4.21. APC2 genine ait, FAM ile işaretlenmiş real time PCR amplifikasyon eğrisi. 48 Hastaların distal kolonlarının metilasyon durumu ile ekspresyonları karşılaştırıldığında; SFRP2 için, distal kolonda metile olan19 kişinin ekspresyonlarının ortalaması 1,42±0.41 iken unmetile olan 1 kişinin ekspresyonu 0,78±0,41 bulundu. Proksimal kolonda ise; metile olan 17 kişinin ekspresyon ortalaması 1,47±0,45, unmetile olan 3 kişinin ortalaması 1,07±0,57 bulundu. SFRP4 geninde distal kolonda metile olan 19 kişinin ekspresyonlarının ortalaması 1,11±0.35 iken unmetile olan 1 kişini ve ekspresyonu 2,43±0,35 bulundu. SFRP4 için proksimal kolonda metile olan 12 kişinin ekspresyon ortalaması 0,93±0,27, unmetile olan 8 kişinin ortalaması 1,10±0,70 bulundu. Bu fark istatistiksel olarak anlamlı değildi (p=0,468). SFRP5 geni için distal kolonu metile olan 12 kişinin ekpresyon ortalaması 0,93±0,51, unmetile olan 8 kişinin ekspresyon ortalaması 1,41±0,26 olup metile olanların ekspresyonu azalmıştı ve bu fark istatistiksel olarak anlamlı (p=0,015) bulundu. SFRP5 proksimal kolonda metile olan 13 kişinin ortalaması 0,90±0,42 unmetile olan 7 kişinin 1,26±0,40 olarak bulundu. Bu ilişki istatistiksel olarak anlamlı değildi (p=0,080). APC1 distal kolon için metile olan 4 kişinin ekpresyon ortalaması 3,28±4,48 ekspresyonu değerlendirilebilen unmetile olan15 kişinin ortalaması 2,98±3,70 bulundu. Proksimal kolonda ekspresyonu değerlendirilebilen 9 metile kişinin ortalaması 2,84±3,58 unmetile olan 9 kişinin ekspresyon ortalaması ise 0,72±0,23 ve bu ilişki istatistiksel olarak anlamlı değildi (p=0,114). APC2 için distal kolonda metile olan ekpresyonu değerlendirilebilen 7 kişinin ortalaması 1,19±0,48 unmetile olup ekspresyonu değerlendirilebilen 11 kişinin ortalaması 1,44±0,91 (p=0,462) bulundu ve bu fark istatistiksel olarak anlamlı değildi. APC2 proksimal kolonda ekspresyonu değerlendirilebilen metile 5 kişinin ortalaması 1,35±0,45 unmetile olan 14 kişinin ekspresyon ortalaması ise 1,20±0,68 olarak bulundu. Hasta ve kontrol gruplarının proksimal ve distal kolonundan alınan örneklerinin SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerin arasında ekspresyon düzeyleri arasında bir ilişkiye rastlanmadı (Çizelge 4.2). Hasta ve kontrollerin gruplarında bazı genlerde çalışılan kişi sayısından daha az kişide ekspresyon bulgusu elde edildi. 49 Çizelge 4.2. Ülseratif kolit hastaları ile kontrol gruplarının proksimal ve distal kolonlarından alınan örneklerin SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin ekspresyon oranı (E) değerinin ortalamaları ve standart sapmaları. D:Distal, P: Proksimal GEN APC1_D E değeri Ortalaması Standart Sapma Standart Hata 19 3,04332 3,806595 0,873293 kontrol 14 1,81029 2,405780 0,642972 18 1,78578 2,698554 0,636055 kontrol 14 0,87314 0,528305 0,141196 1,34433 0,764438 0,180180 GRUP N hasta p Değeri 0,296 APC1_P hasta 0,224 APC2_D hasta 18 0,311 kontrol 14 APC2_P 1,09536 0,542992 0,145121 19 1,24479 0,625936 0,143599 kontrol 14 1,10979 0,269475 0,072020 20 1,39150 0,424981 0,095029 kontrol 14 1,14579 0,319665 0,085434 1,13605 0,461128 0,103111 hasta 0,408 SFRP2_D hasta 0,077 SFRP2_P hasta 20 0,645 kontrol 14 SFRP4_D 1,07321 0,245333 0,065568 20 1,17850 0,457725 0102350 kontrol 14 1,08629 0,291176 0,077820 20 1,00130 0,485713 0,108609 kontrol 14 0,96414 0,207596 0,055482 1,12490 0,487984 0,109117 hasta 0,511 SFRP4_P hasta 0,790 SFRP5_D hasta 20 0,613 kontrol 14 SFRP5_P 1,04729 0,345501 0,092339 20 1,03080 0,439783 0,098338 kontrol 14 0,99764 0,215682 0,057643 hasta 0,773 50 Hastaların distal kolonlarında inflamasyon durumu (hafif/ şiddetli) ile distal kolonda SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin ekspresyon düzeyleri arasında anlamlı bir ilişki bulunmadı (Çizelge 4.3). Çizelge.4.3. Hastaların distal kolon örneklerinde inflamasyonu ile SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin ekspresyon oranı (E) değerinin ortalamaları ve standart sapmaları. Gen APC1 APC2 SFRP2 SFRP4 SFRP5 İnflamasyon Durumu N Ortalama Standart Sapma Standart Hata hafif 7 3,39000 3,559290 1,345285 şiddetli 12 2,84108 4,084111 1,178981 hafif 7 1,29314 0,484805 0,183239 şiddetli 11 1,37691 0,921628 0,277881 hafif 7 1,56800 0,487312 0,184187 şiddetli 13 1,29646 0,373188 0,103504 hafif 7 1,33357 0,421877 0,159455 şiddetli 13 1,09500 0,470273 0,130430 hafif 7 1,09214 0,632811 0,239180 şiddetli 13 1,14254 0,419344 0,116305 p Değeri 0,771 0,828 0,180 0,278 0,832 51 5. TARTIŞMA Bu çalışmada sürveyans kolonoskopisi yapılan distal tip ÜK tanısı alan hastalar ile kanser taraması yapılan sağlıklı kişilerin proksimal ve distal kolonlarından alınan örneklerde Wnt yolağında etkili olan genlerden SFRP2, SFRP4, SFRP5, APC1 ve APC2 genlerinin metilasyon durumları ile ekspresyon düzeyleri incelendi. Hasta ve kontrollerin distal ve proksimal kolon örneklerinin SFRP2 geninin metilasyon durumları arasında istatistiksel olarak anlamlı bir fark bulunmadı. SFRP2 genin ekspresyonlarının proksimal kolon örneklerinde istatistiksel anlamlı bir fark bulunmazken hasta grubunun distal kolon örneklerinde ekspresyon oranları 1,39±0,42; kontrol grubunda ekspresyon oranları 1,14±0,32 bulundu. Hastaların distal ve proksimal kolonlarından alınan örneklerin metilasyon durumu ve ekpresyonları arasında SFRP2 geni için istatistiksel olarak anlamlı bir fark bulunmadı. SFRP4 geni incelendiğinde hasta ve kontrollerin proksimal kolon örneklerinde metilasyon durumunda istatistiksel olarak anlamlı bir fark vardı (p=0,018) ve proksimal kolonda metilasyon azalmış görünüyordu. SFRP4 genin hastaların distal ve proksimal kolonlarından alınan örneklerde metilasyon durumu ve ekpresyon oranları arasında anlamlı bir ilişki gözlemlenmedi. SFRP5 geni için hastaların distal kolonlarından alınan örneklerin metilasyon durumu ve ekpresyon oranları arasında istatistiksel olarak anlamlı bir ilişki bulundu (p=0,015). Distal kolonda SFRP5 geninin metilasyonu arttıkça ekspresyonunun azaldığı görüldü. APC2 geni için hastaların distal kolonundan alınan örnekler ile kontrollerin metilasyon durumu arasında istatistiksel olarak anlamlı bir fark vardı (p=0,018). Hastaların distal kolonunda kontrollere göre metilasyon artmıştı. İnflamasyon durumu ile bu genlerin metilasyonu ve ekspresyonu arasında istatistiksel olarak anlamlı bir ilişki bulunmadı. İBH’da artmış mortalitenin asıl sebebi KRK’dır. Bu risk hastalığa maruziyet süresiyle artmaktadır. Bu nedenle uzun süredir bu hastalığa sahip kişilere 1-3 yıl arayla düzenli sürveyans kolonoskopileri önerilmektedir. Ancak İBH ilişkili karsinomların kolonoskopide tanınması zordur ve displazi %90 güvenlikte bulmak için kolonoskopik biyopsi sayısını 33 ve üzerine çıkarmak gerekmektedir. Sürveyansın etkinliğini arttırmak için uzun süreli hastalıktan muzdarip hastalar arasında İBH ilişikili neoplastik lezyonların erkenden saptanması için objektif ve güvenli moleküler belirteçlere büyük 52 ihtiyaç vardır. Kanser ilişkili genlerin DNA metilasyon bağımlı sessizleşmesi KRK’da önemli ve erken bir bulgudur. p16, E-cadherin, hMLH1, p14, HPP1, ER vb gibi genlerin kanser spesifik metilasyonları İBH ilişkili neoplazi durumunda (displazi ve kanser) varolduğu bildirilmiştir (10-14). Normalde Wnt sinyal yolağı hücre dışında Wnt ligandlarını bağlayan SFRP’ler, WIF1 ve LRP yüzey molekülünü bağlayan DKK ailesi proteinleri tarafından inhibe edilir. Hücre içinde ise APC, aksin, GSK3 kompleksi tarafından β-katenin degredasyonu ile hedef genlerin ekspresyonları inhibe edilir. SFRP’ler, WIF1ve DKK proteinleri ve APC’nin promotor metilasyonu ile inaktive olmaları sonucu kanser hücresinde Wnt sinyalinin aktivasyonu ve genlerin ekspresyonları artar. APC promotor metilasyonu KRK’da sağ kolonda sol kolona göre daha fazla bulunduğu ve hata tamir eksikliği de sağ kolonda daha fazla olduğu bildirilmiştir (115). Prasad CP ve ark. (116) invaziv duktal karsinomda E-CD ve APC protein kaybı ve β-katenin/Wnt sinyal yolağı aktivasyonu ile CDH1 ve APC promotor metilasyonunun ilişkisini göstermişlerdir. Bu çalışmada APC1genin promotor metilasyonu ve ekspresyonu arasında istatistiksel olarak anlamlı bir fark bulunmadı. Tümörlerde hipermetilasyon ile inhibitör Wnt ligandları olan SFRP’lerin susturulduğu bilinmektedir (117). SFRP2 metilasyon sıklığını kolorektal kanser (118) ve Zou ve ark. (109) barret özafagusunda, karsinojenik transformasyon yoluyla metilasyon sıklığının arttığı veya varolan özafagus kanserinde metilasyonlarının arttığını bildirmişlerdir. Urakami S ve ark. (110) mesane tümörünün progresyonu ve patogenezi ile ilgili çalışmada Wnt antagonist genleri SFRP1, SFRP2, SFRP4, ve SFRP5, DKK3, ve WIF1 genleri metilasyon düzeylerini yüksek ve mesane tümöründe MSP negatif gruba göre MSP pozitif grupta mRNA ekspresyon düzeyleri oldukça yüksek bulmuşlardır. promotor Onların bu sonuçları mesane tümörünün patogenezini içeren hipermetilasyonu yoluyla Wnt antogonistlerinin zayıflatılmasına neden olduğunu desteklemektedir. fonksiyonlarının Urakami S ve ark.’nın (110) çalışmalarında idrar örneklerinden çıkan sonuçlar tümör doku DNA ile benzer ve metilasyonun yüksek yüzdesini göstermiştir. Ayrıca normal olan kontrollerde idrar DNA’sında %90’dan fazla anormal metilasyon olmadığını bulmuşlardır. Bu bulgular (a)Wnt antagonist genlerinin metilasyonun bulunmasının makul ve güvenilir olduğu ve (b) Wnt antagonist genlerinin idrar metilasyon skoru, mesane tümörü için mükemmel bir noninvaziv tanı biyomarkırı olarak kullanılabileceğini göstermiştir. Sonuç olarak, 53 Wnt antagonist genlerinin hipermetilasyonu mesane tümörünün patogenezinde önemli bir rol oynadığı ve mesane tümörlü hastaların idrarında kolaylıkla saptanabileceğini bildirmişlerdir. Fakat Dulaimi ve ark. (119) sistitli hastaların idrar DNA’sında tümör süpresör genler APC, RASSF1A, p14’ün metilasyon olmadığını gözlemlediklerini bildirmişlerdir. Wang DR ve ark. (120) 2008’te yaptıkları çalışmada doku ve fekal DNA SFRP2 geninin metilasyonu karşılaştırmışlar ve dokuda KRK’da % 92,1, adenomda %74,3 ve hiperplastik polipte %53,8 ve aynı vakaların fekal örneklerinde sırasıyla %87, %61,8 ve %42,3 hipermetillenmiş olduğunu bildirmişlerdir. Buna göre; dışkı örneklerinden fekal DNA izole edildiğinde, SFRP2 geninin metilasyonu kolorektal kanser için taramada moleküler bir biyomarkır olarak kullanılabileceğini bildirmişlerdir. Suzuki H ve ark. (118) mutant β-katenin veya APC yoluyla Wnt sinyal aktivasyonu SFRP’lerin aşırı ekspresyonu ile kısmen baskılanabileceğini bildirmişlerdir. Cheng YY ve ark. (121) gastrik kanserde SFRP1, SFRP2, SFRP4 ve SFRP5 mRNA ekspresyon düzeyleri ve metilasyon durumlarını çalışmışlar ve bunlardan sadece SFRP2’nin gastrik kanserde sessizleştiğini ve metillendiğini bulmuşlardır. SFRP2 promotor metilasyonunu özellikle gastrik kanserde mRNA ekspresyonu düşük veya yokluğu ile ilişkili bulmuşlardır. Ayrıca gastrik kanser hücre hatlarını demetilleyici ajan 5-aza-20-deoksisistidin ile tedaviden sonra SFRP2 ekspresyonunun geri döndüğünü teyit etmişler ve bu da SFRP2 promotor bölgesinin hipermetilasyonunun gastrik kanserde SFRP2 transkripsiyonel sessizleşmesi için kritik olabileceğini göstermiştir. Hipermetilasyon mide kanserinde %73,3, intestinal metaplazide %37,5 ve komşu kanser dışı dokularda %20 olarak bulmuşlardır. Bu nedenle SFRP2 ‘deki epigenetik değişiklikler malign transformasyona yol açan önemli bir mekanizma olduğunu mide kanseri olmayan hastalarda bile pre-neoplastik evrede olduğunu destekler. Bu veriler birlikte ele alındığında bir tümör süpresör gen olarak SFRP2’nin gastrik kanserde etkisi olduğunu ve sessizleşmesi tümör büyüme ve genişlemesini arttırabileceğini göstermişlerdir (121). Takagi H ve ark. (122) karaciğer kanser hücre hatlarında SFRP4’ün daha az sıklıkla metillendiğini SFRP1, SFRP2 ve SFRP5’in ise daha fazla metillendiğini göstermişlerdir. Ayrıca RT-PCR analizi ile metillenmiş hücre hatlarında SFRP1, SFRP2 ve SFRP5 kayıp ya da downregüle olduğunu ve bir DNA metil inhibitörü 5-aza-20-deoksisistidin ile tedaviden sonra ekspresyonların düzeldiğini göstermişlerdir. SFRP genlerini hepatosüllüler kanserde 54 (HSK) epigenetik sessizleşmesinin yaygın bir bulgu olduğunu bildirmişlerdir. Ayrıca, SFRP’lerin metilasyonu hepatokarsinogenezlerde preneoplastik bu metilasyonun lezyonlarda sık oldukça erken görüldüğünü evrede ve çıktığını bildirmişlerdir. Viral enfeksiyonlarla ilişkilendirilmiş siroz ve kronik hepatitin preneoplastik lezyonlar olduğu düşünülmektedir. SFRP genlerinin metilasyonu ve viral enfeksiyonlar arasındaki mekanik bağlantıyı açıklamak için daha fazla çalışmaya ihtiyaç olmasına rağmen metilasyonun tespiti HSK gelişimi yüksek riskli hastaların takibi için yararlı moleküler markır olabileceğini bildirmişlerdir (122). Biz de yaptığımız çalışmada SFRP2 genin ekspresyonunu distal kolon örneklerinde hasta grubu ile kontrol grubu arasında istatistiksel olarak anlamlı olmayan bir fark gözlemledik (distal SFRP2 hasta 1,391±0,424; distal SFRP2 kontrol 1,145±0,319). Hastaların ve kontrollerin distal kolonlarından alınan örneklerin SFRP2 geni için metilasyon durumu arasında istatistiksel olarak anlamlı bir ilişki bulunmamıştır (p=0,385). SFRP4 genini incelendiğimizde ise hasta ve kontrollerin proksimal kolon örneklerinde metilasyon durumunda istatistiksel olarak anlamlı bir fark vardı (p=0,018) ve hastaların proksimal kolon örneklerinde kontrollere göre metilasyon azalmış görünüyordu. SFRP4 genin hastaların distal kolonlarından alınan örneklerin metilasyon durumu ve ekpresyonları arasında ise anlamlı ilişki saptanmamıştır. SFRP5 geni için hastaların distal kolonlarından alınan örneklerin metilasyon durumu ve ekpresyonları arasında istatistiksel olarak anlamlı bir ilişki gözlemlenmiştir (p=0,015). Distal kolonda SFRP5 metilasyonu arttıkça ekspresyonun azaldığı gözlemlenmiştir. Distal kolonda hasta ve kontrollerin metilasyon durumlarının APC2 geni için karşılaştırıldığında anlamlı bir fark bulunmuştur (p=0,018). Hasta grubunda kontrol grubuna göre metilasyon daha fazla olduğu gözlemlenmiştir. İnflamasyon mikrobiyal infeksiyonlarda, alerjenlere ve toksik kimyasallara maruziyetle, otoimmün hastalıkla ve obezite içeren birçok etkenle ortaya çıkabilir. Dengeli bir inflamatuvar yanıtın antitümorejenik etkisi olabilir; ancak devam eden veya kronik inflamatuvar yanıt, immün yanıt artık bozulmuş olduğundan genellikle zararlı hale gelmektedir. Kronik inflamasyonla kanser arasındaki nedensel ilişki oldukça iyi bilinmektedir ve gastrointestinal kanalın birçok kronik olarak inflame organı bu ilişkiyi gösterir. Buna inflamatuvar barsak hastalığı iyi bir örnek olup genel nüfüsa göre 55 kolorektal kanser riski 2-3 kat artmıştır. Kolit ilişkili kanserin çok yönlü olduğu düşünülmektedir ve muhtemelen genetik faktörlerin, çevresel faktörlerin, hastalığın süresinin, yaygınlığın ve şiddetinin kombinasyonu kolit ilişkili kanserin gelişimine katkıda bulunduğu düşünülmektedir. Son zamanlarda DNA metilasyonundaki özel değişikliklerin; epigenetik değişiklikler, inflamasyon sırasında ve inflamasyon ilişkili kanser sırasında meydana geldiği bildirilmiştir. Mukozal inflamasyonun global metabolik sonuçlarının incelendiği bir çalışmada hücresel metilasyon reaksiyonları ile ilişkili spesifik metabolit düzeylerinin inflamasyona yanıtta belirgin olarak değiştiği, ayrıca tüm hücresel metilasyonun kilit enzimlerinin ekspresyonunun arttığı da gösterilmiştir. (123). Hücresel metilasyonun inhibisyonu hastalık alevlenmesiyle sonuçlanmıştır ve metilasyonu uyaran folat idamesi kolitin kısmen iyileşmesi ile sonuçlanmıştır. Tüm metilasyondaki global bu bulgular değerlendirildiğinde değişiklikleri gösteren bu inflamasyon sonuçlar sırasında mukozal epitelde metilasyonun koruyucu bir rolü olduğu anlamına gelir. Folatın yararlı etkisi metilasyonun global kaybının önlenmesi ve inflamasyonun baskılaması yeteneği nedeniyle olduğu düşünülmektedir. (124). İBH ilişkili neoplazide Wnt sinyal yolağının ilk epigenetik katılımının gösterildiği bir çalışmada Wnt sinyal yolağının genlerin metilasyonu uzun süredir IBH kolitli olan hastalarda daha önce başladığı ve IBH ilişkili neoplazi geliştirilmesi sırasında kademeli olarak bu genlerin metilasyonunun arttığı bulunmuştur (98). Dhir M ve ark.’nın (98) yaptığı çalışmada %29’u metillenen SFRP2 ve %7’si metillenen WIF1 hariç tüm genler için metilenmeme normal kolon örnekleri ile eşit bulunmuştur. Tüm Wnt sinyal yolağının genlerinin (LKB1 hariç) metilasyonunun İBH ilişkili kolit ve İBH ilişkisiz neoplazili örneklerde sık görüldüğü bildirilmişitr. İBH kolitli örneklerde görülen Wnt sinyal yolağının genlerinin metilasyonun sıklığı normal kolonda da görülenden anlamlı olarak daha yüksek bulunduğu fakat SFRP2’de fark istatistiksel olarak anlamlı bulunmadığı bildirilmiştir (98). Ayrıca Wnt sinyal yolağı genlerin (tüm İBH ilişkili neoplazilerde unmetile olan LKB1 hariç) metilasyon sıklığı İBH kolit örnekler ile karşılaştırıldığında İBH ilişkili neoplazili örneklerde arttığı gösterilmişlerdir. APC1A, APC2, SFRP1 ve SFRP2’nin metilasyonu İBH kolit ile karşılaştırıldığında İBH ilişkili neoplazide anlamlı olarak yüksek bulmuşlar (sırasıyla APC1A %43.8 ile %16.0, p=0.05; APC2 %81.3-%48.0, p=0.03; SFRP1 %87.5-%54.1, 56 p=0.03 ve SFRP2 %86.7-%52.0, p=0.03). Ayrıca tüm Wnt sinyal yolağı genlerinin metilasyonunu İBH ilişkili kanserlerle uyan evrede (Evre 1 ve 2) olan 24 sporadik KRK de test edilmişler ve sporadik KRK’de görülen Wnt sinyal yolağındaki genlerin metilasyon sıklığı SFRP4 ve WIF1 dışında İBH ile ilişkili neoplaziler (displazileri ve kanser dahil) görülenlere benzer bulunmuştur. SFRP4’ün, sporadik KRK’lere (%37,5) göre İBH ilişkili kanserlerde (%81,3) önemli ölçüde daha fazla metillendiğini göstermişlerdir. Aksine WIF1 İBH ile ilişkili neoplazilere (%68,8) göre sporadik KRK’lerde (%100) önemli ölçüde daha fazla metillenmiş olduğunu bulmuşlardır (98). Bizim çalışmamızda gözlemlediğimiz SFRP4’ün metilasyonunun azaldığı bulgusu Dhir M ve ark.’nın (98) bulgusu ile benzer değil iken Takagi ve ark.'nın (122) karaciğer kanser hücre hatlarında SFRP4’ün daha az sıklıkla metillendiği bulgusu ile benzerdir. Erken dönemdeki bu metilasyon değişikliklerinin malignite riski açısından ne ifade ettiğini söyleyebilmek çalışma grubumuzdaki prenoplastik/neoplastik olguların olmaması nedeniyle mümkün değildir. Yine aynı çalışmada (98) İBH ile ilişkili displazide APC2, SFRP1, SFRP2 ve APC1A’in erken metilasyon bulguları bu lezyonların tanısında daha güvenilir bir belirteç olabileceği bildirilmiştir. Aslında, düşük dereceli displazi ve yüksek dereceli displazi olan tüm hastalar displazi için bir belirteç olarak kullanılabileceği düşünülen APC2 %100 metilasyon gösterdiği de bulunmuştur. Çalışmamızda APC2 geni için hastalar ile kontrollerin distal kolonlarında metilasyon durumu arasında istatistiksel olarak anlamlı bir fark vardı ve İBH’lı hastalarda metilasyonun arttığı gösterilmiştir. APC2, yakın zamanlarda tanımlanmış, APC1A ile yüksek homolojisi gösteren bir gendir (125) APC2’nin, APC1A gibi Wnt sinyalini modüle de edebileceği bildirilmiştir (125, 126). APC1A da mutasyonlar dışında İBH ile ilişkili neoplazilerde diğer tümör baskılayıcı gen olan p53’ün mutasyonunun zamanlaması ve sıklığı arasında farklılık gösterir. P53 mutasyonları sporadik KRK’de geç ortaya çıkar. p53 mutasyonları İBH ilişkili neoplazi de erken ve daha sık görülürken kanserler adenomlarla karşılaştırıldığında daha sık görülür. Düşük dereceli displazilere benzer erken lezyonlar ülseratif kolit ve hatta bazen displastik olmayan mukozada P53 mutasyonuna sahip olabilir (73,75). İlginç bir şekilde, APC2 bir anti-apoptotik gen Bcl2 ve p53 ile sırayla etkileşen 53BP2 bir protein ile etkileşimi gösterilmiştir. Bu hücre döngüsü sürecinin ve hücre ölümünün p53/Bcl2-bağımlı yolakta APC2’nin işlevsel bir rolünü düşündürmektedir (127). İBH ile ilişkili neoplazi incelendiğinde ise P53 yolağı 57 ile APC2’nin etkileşimi çok ilginçtir. Erken displastik lezyonlar P53 mutasyonu ve Bcl2 aşırı ekspresyonunun yüksek seviyesini gösterir (74,75,128). Benzer şekilde, APC2’nin inflame kolon lezyonlarında yüksek seviyelerde metile olduğunu bulunmuştur. Bizim çalışmamızda da APC2’i inflame kolon da metillenmiş olarak bulundu. Fakat çalışma grubumuzda preneoplastik, neoplastik lezyon olmadığından dolayı neoplazi gelişimindeki rolü hakkında bir yorum yapmak zordur. Bu nedenle, APC2 metilasyonu, p53 mutasyonunun ve Bcl2 aşırı ekspresyonunun bu lezyonların patogenezinde sinerjik bir rol oynayabiliceği düşünülmektedir. Bu, bulgunun daha fazla araştırılması gerektirmektedir. 58 6. SONUÇLAR ve ÖNERİLER 1. Wnt sinyal yolağı genlerinin metilasyonu ÜK hastalarında bildirilmiştir. Wnt sinyal yolağı genlerinin metilasyon sıklığının İBH ilişkili neoplazi gelişimi sırasında arttığını bildiren çalışmalar olmakla birlikte bizim çalışmamızda preneoplastik lezyon ve neoplazi olmaması nedeniyle böyle bir ilişkiyi çalışalamadı. 2. İBH ile ilişkili inflamasyonda, APC2’nin artmış metilasyon bulguları ve SFRP5’in metilasyon artışı ile ekspreyonunun azalması ve SFRP4’de metilasyon azlığının ÜK’te inflamasyona eşlik ettiği bulgusunun kolit ile ilişkili neoplazinin erken tespitinde kullanılabilecek yöntem ve objektif belirteç sağlayıp sağlayamayacağının araştırılması gerekimektedir. 3. Mevcut bulguların immünohistokimyasal çalışmalarlada da desteklenmesi bulguların değerini daha da arttıracaktır. 4. İBH’da KRK’in önlenmesi için stratejiler patoloğun uzmanlığından bağımsız olan displazi ve kanser için objektif belirteçlerin eklenmesi ile güçlendirilebilir. 59 7. KAYNAKLAR 1. Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer: a population-based study. N Engl J Med 1990;323:1228-1233. 2. Langholz E, Munkholm P, Davidsen M, Binder V. Colorectal cancer risk and mortality in patient with ulcerative colitis. Gastroenteology 1992;103:1444-1451. 3. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001;48(4):526–535. 4. Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, Sommers SC, Yardley JH. Dysplasia in inflammotory bowel disease: standardized classification with provisional clinical applications. Hum Pathol 1983;14:931-968. 5. Kornbluth A, Sachar DB. Practice Parameters Committee of the American College of Gastroenterology: Ulcerative colitis practice guidelines in adults (update): American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol 2004;99:1371-1385. 6. Farrell RJ, Peppercorn MA. Ulcerative colitis. Lancet 2002;359(9303):331–340. 7. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000;16(4):168–174. 8. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005;2(Suppl 1):S4–S11. 9. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003;349(21):2042–2054. 10. Azarschab P, Porschen R, Gregor M, Blin N, Holzmann K. Epigenetic control of the Ecadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer 2002;35(2):121–126. 11. Fleisher AS, Esteller M, Harpaz N, Leytin A, Rashid A, Xu Y, Liang J, Stine OC, Yin J, Zou TT, Abraham JM, Kong D, Wilson KT, James SP, Herman JG, Meltzer SJ. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res 2000;60(17):4864–4868. 60 12. Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res 1998;58(17):3942–3945. 13. Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 2001;61(9):3573–3577. 14. Moriyama T, Matsumoto T, Nakamura S, Jo Y, Mibu R, Yao T, Iida M. Hypermethylation of p14 (ARF) may be predictive of colitic cancer in patients with ulcerative colitis. Dis Colon Rectum 2007;50(9):1384–1392. 15. de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci 2007;12:471–491. 16. Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res 2005;306(2):357–363. 17. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992;1(4):229–233. 18. 932. Potter JD. Colorectal cancer: molecules and populations. J Natl Cancer Inst 1999;91(11):916– 19. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997;11(24):3286–3305. 20. Klingensmith J, Nusse R, Perrimon N. The Drosophila segment polarity gene dishevelled encodes a novel protein required for response to the wingless signal. Genes Dev 1994;8(1):118–130. 21. Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996;382(6592):638– 642. 22. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998;281(5382):1509– 1512. 61 23. Howe LR, Subbaramaiah K, Chung WJ, Dannenberg AJ, Brown AM. Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res 1999;59(7):1572–1577. 24. Stutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999;96(10):5522-5527. 25. Bafico A, Gazit A, Pramila T, Finch PW, Yaniv A, Aaronson SA. Interaction of frizzled related protein (FRP) with Wnt ligands and the frizzled receptor suggests alternative mechanisms for FRP inhibition of Wnt signaling. J Biol chem 1999;274(23):16180-16187. 26. Zorn AM. WNT signaling: antagonistic Dickkopfs. Curr Biol 2001;11(15):R592-R595. 27. Tarmin L, Yin J, Harpaz N, Kozam M, J, Noordzij Antonio LB, Jiang HY, Chan O, Cymes K, Meltzer SJ. Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer res 1995;55(10)2035-2038. 28. You J, Nguyen Av, albers CG, Lin F, Holcombe RF. Wnt pathway-related gene expression in inflammatory bowel disease. Dig Dis Sci 2008;53(4):1013-1019. 29. You XJ, Bryant PJ, Jurnak F, Holcombe RF. Expression of Wnt pathway components frizzled and disheveled in colon cancer arising in patients with inflammatory bowel disease. Oncol rep 2007;18(3):691-694. 30. van Dekken H, Wink JC, Vissers KJ, Franken PF, Ruud Schouten W, Hop WCJ, Kuipers EJ, Fodde R, Janneke van der Woude C. Wnt pathway-related gene expresion during malignant progression in ulcerative colitis. Acta Histochem 2007;109(4):266-272. 31. Aust DE, Terdiman JP, Willenbucher RF, Chang CG, Molinaro-Clark A, Baretton GB, Loehrs U, Waldman FM. The APC/beta catenin pathway in ulcerative colitis related colorectal carcinomas: a mutational analysis. Cancer 2002;94(5):1421-1427. 32. Aust DE, Terdiman JP, Willenbucher RF, Chew K, Ferrell L, Florendo C, Molinaro-Clark A, Baretton GB, Löhrs U, Waldman FM. Altered distribution of beta-catenin, and its binding proteins E-cadherin and APC, in ulcerative colitis related colorectal cancers. Mod Pathol 2002;14(1):29-39. 33. Feoglio-Preiser CM, Noffsinger AE, Stemmermann GN, Lantz PE, Listrom MB, Rilke FO. Inflamatory Bowel Disease. In: Norvi el M, ed. Gastrointestinal Pathology An Atlas and Text. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 1999. p.659-716. 62 34. Rosai J. Colitis. In: Houston M, ed. Rosai and Ackerman’s Surgical Pathology. 9th ed. Philadelhia: Elsevier; 2004. p.782-8. 35. Owen DA, Kelly JK. Disease of the large bowel. In: Owen DA, Kelly JK, eds. Atlas of Gastrointestinal Pathology. 1st ed. Philadelphia: W.B. Saunders Company; 1994. p.141-2 36. Judge TA, Lichtenstein GR Inflammatory Bowel Disease. In: Friedman SL, McQuaid KR, Grendell JH, eds. Current diagnosis and treatment in gastroenterology. 2nd ed. New York; Lange Medical Boks/McGraw- Hill: 2003. p.108-30. 37. Ming S, Goldman H. Ulcerative colitis and Crohn’s Disease. In: Ming S, Goldman H, eds. Pathology of the Gastrointestinal Tract. 1st ed. Philadelphia: W.B. Saunders Company; 1992.p.643-63. 38. Odze R. Diagnostic problems and advances in inflammatory bowel disease. Mod Pathol 2003;16(4):347-58. 39. Özin YÖ, Ülker A. İnflamatuvar bağırsak hastalıkları. In: Özden A, Şahin B, Yılmaz U, Soykan İ, eds. Gastroentereloji. 1. baskı, İstanbul, Fersa Matbaacılık Ltd. Şti. 2002. p.283-7. 40. Sinclair TS, Brunt PW, Mowat NA. Nonspecific proctocolitis in northeastern Scot land: a community study. Gastroenterology 1983;85 (1):1-11. 41. 315 Edwards Fc and Truelo SC. The Course And Prognosis of Ulcerative Colitis. Gut 1963;4:299- 42. Tözün N, Altuğ Ö. İltihabi Barsak Hastalıkları. In: Memik F, ed. Klinik Gastroenteroloji. 1.baskı, İstanbul, Nobel Tıp Kitapevleri Ltd Şti. Nobel Güneş tıp kitapevi. 2004. p.448-61. 43. Su C, Lichtenstein GR. Ulcerative Colitis. In: Feldman M, Friedman LS, Brandt LJ. Sleisenger MH, eds. Sleisenger Ford tran’s Gastrointestinal and liver disease. 8th ed. Philadelphia; Saunders Elseiver: 2006. p.2499-548. 44. Silverberg SG, Delellis RA, Frable WJ. Non-Neoplastic Diseases of the Small and Large Intestine. In: Silverberg SG, Delellis RA, Frable WJ, eds. Principles and Practice of Surgical Pathology and Cyto pathology. 3rd ed. New York: Churchill Living stone; 1997. p.1780-3. 45. Dixon MF, Brown LJ, Gilmour HM, Price AB, Smeeton NC, Talbot IC, Williams GT. Observer variation in the assessment of dysplasia in ulcerative colitis. Histopathology. 1988 Oct;13(4):385-97. 63 46. Sharan R, Schoen RE. Cancer in inflammatory bowel disease. An evidence-based analysis and guide for physicians and patients. Gastroenterol Clin North Am 2002;31(1):237-54. 47. Eaden JA, Mayberry JF. Colorectal cancer complicating ulcerative colitis: a review. Am J Gastroenterol 2000;95(10):2710-9 48. Greenstein AJ, Sachar DB, Smith H, Pucillo A, Papatestas AE, Kreel I, Geller SA, Janowitz HD, Aufses AH Jr. Cancer in universal and left-sided ulcerative colitis: factors determining risk. Gastroenterology 1979; 77(2):290-4. 49. Mir-Madjlessi SH, Farmer RG, Easley KA, Beck GJ. Colorectal and extra colonic malignancy in ulcerative colitis. Cancer 1986;58(7):1569-74. 50. Prior P, Gyde SN, Macartney JC, Thompson H, Water house JA, Allan RN. Cancer morbidity in ulcerative colitis. Gut 1982;23(6):490-7. 51. Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a populati on-based study. Cancer 2001;91(4):854-62. 52. Hendriksen C, Kreiner S, Binder V. Longterm prognosis in ulcerative colitis-based on results from a regional patient group from the county of Copenhagen. Gut 1985;26(2):158-63. 53. Stonnington CM, Phillips SF, Zinsmeister AR, Melton LJ 3rd. Prognosis of chronic ulcerative colitis in a community. Gut 1987;28(10):1261-6. 54. Mellemkjaer L, Olsen JH, Frisch M, Johansen C, Gridley G, McLaughlin JK. Cancer in patients with ulcerative colitis. Int J Cancer 1995;60(3):330-3. 55. Gilat T, Fireman Z, Grossman A, Hacohen D, Kadish U, Ron E, Rozen P, Lilos P. Colorectal cancer in patients with ulcerative colitis. A population study in central Israel. Gastroenterology 1988;94(4):870-7. 56. Karlén P, Löfberg R, Broström O, Leijonmarck CE, Hellers G, Persson PG. Increased risk of cancer in ulcerative colitis: a populati on-based cohort study. Am J Gastroenterol; 1999;94(4):1047-52. 57. Shetty K, Rybicki L, Brzezinski A, Carey WD, Lashner BA. The risk for cancer or dysplasia in ulcerative colitis patients with primary sclerosing cholangitis. Am J Gastroenterol 1999;94(6):1643-9. 64 58. Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC. A prospective study of family history and the risk of colorectal cancer. N Engl J Med 1994;331(25):1669-74. 59. Schoen RE. Families at risk for colorectal cancer:risk assessment and genetic testing. J Clin Gastroenterol 2000;31(2):114-2. 60. UllmanTA, Itzkowitz 2011;140:1807–1816. SH. Intestinal Inflammation and Cancer. Gastroenterology 61. Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology 2010;138:746– 774. 62. Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 2004;287:G7–G17. 63. Eaden J, Abrams K, Ekbom A, Jackson E, Mayberry J.. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther 2000;14:145–153. 64. Pinczowski D, Ekbom A, Baron J, Yuen J, Adami HO. Risk factors for colorectal cancer in patients with ulcerative colitis: a case-control study. Gastroenterology 1994;107:117–120. 65. Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004;126:451–459. 66. Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, Bodian C, Ullman T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology 2007;133:1099–1105. 67. Mathy C, Schneider K, Chen YY, Varma M, Terdiman JP, Mahadevan U. Gross versus microscopic pancolitis and the occurrence of neoplasia in ulcerative colitis. Inflamm Bowel Dis 2003;9:351–355. 68. Rubin CE, Haggitt RC, Burmer GC, Brentnall TA, Stevens AC, Levine DS, Dean PJ, Kimmey M, Perera DR, Rabinovitch PS. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992;103:1611–1620. 65 69. Itzkowitz SH. Molecular biology of dysplasia and cancer in inflammatory bowel disease. Gastroenterol Clin North Am 2006;35: 553–571. 70. Willenbucher RF, Aust DE, Chang CG, Zelman SJ, Ferrell LD, Moore DH 2nd, Waldman FM.. Genomic instability is an early event during the progression pathway of ulcerativecolitis-related neoplasia. Am J Pathol 1999;154:1825–1830. 71. Redston MS, Papadopoulos N, Caldas C, Kinzler KW, Kern SE. Common occurrence of APC and K-ras gene mutations in the spectrum of colitis-associated neoplasias. Gastroenterology 1995;108:383–392. 72. Umetani N, Sasaki S, Watanabe T, Shinozaki M, Matsuda K, Ishigami H, Ueda E, Muto T. Genetic alterations in ulcerative colitis-associated neoplasia focusing on APC, K-ras gene and microsatellite instability. Jpn J Cancer Res 1999;90: 1081–1087. 73. Burmer GC, Rabinovitch PS, Haggitt RC, Crispin DA, Brentnall TA, Kolli VR, Stevens AC, Rubin CE. Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology 1992;103:1602–1610. 74. Yin J, Harpaz N, Tong Y, Huang Y, Laurin J, Greenwald BD, Hontanosas M, Newkirk C, Meltzer SJ. p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993;104:1633–1639. 75. Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, Burmer GC. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994;107:369–378. 76. Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi AJ, Harris CC. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 2000;60:3333–3337. 77. Sato F, Harpaz N, Shibata D, Xu Y, Yin J, Mori Y, Zou TT, Wang S, Desai K, Leytin A, Selaru FM, Abraham JM, Meltzer SJ. Hypermethylation of the p14(ARF) gene in ulcerative colitisassociated colorectal carcinogenesis. Cancer Res 2002;62:1148–1151. 78. Kohonen-Corish MR, Daniel JJ, te Riele H, Buffinton GD, Dahlstrom JE. Susceptibility of Msh2-deficient mice to inflammation-associated colorectal tumors. Cancer Res 2002;62:2092–2097. 66 79. Cooper HS, Everley L, Chang WC, Pfeiffer G, Lee B, Murthy S, Clapper ML. The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodiuminduced colitis. Gastroenterology 2001; 121:1407–1416. 80. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–612. 81. Qiu W, Wang X, Leibowitz B, Liu H, Barker N, Okada H, Oue N, Yasui W, Clevers H, Schoen RE, Yu J, Zhang L. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc Natl Acad Sci USA 2010;107:20027–20032. 82. Shaker A, Rubin DC. Intestinal stem cells and epithelial-mesenchymal interactions in the crypt and stem cell niche. Transl Res 2010;156:180–187. 83. Shaker A, Swietlicki EA, Wang L, Jiang S, Onal B, Bala S, DeSchryver K, Newberry R, Levin MS, Rubin DC. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J Clin Invest 2010;120:2081–2093. 84. Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science (Wash. DC), 1999;286: 481–486. 85. Kass SU, Wolffe AP. DNA methylation, nucleosomes and the inheritance of chromatin structure and function. Novartis Found. Symp. 1998;214: 22–35. 86. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat. Genet. 1999;21:163–167. 87. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004;,20: 781–810. 88. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17(1): 9–26. 89. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469–480. 90. Polakis P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007;17:45–51. 67 91. He X, Saint-Jeannet JP, Wang Y, Nathans J, Dawid I, Varmus H. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science. 1997; 275:1652–1654. 92. Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4(4):115. 93. Verkaar F, Blankesteijn WM, Smits JF, Zaman GJ. Beta-Galactosidase enzyme fragment complementation for the measurement of Wnt/β-catenin signaling. FASEB J. 2010;24:1205–1217. 94. Verkaar F, Zaman GJ. A model for signaling specificity of Wnt/Frizzled combinations through co-receptor recruitment. FEBS Lett. 2010;584: 3850–3854. 95. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. 96. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat.Rev. Mol. Cell Biol. 2009;10:468–477. 97. Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Exp. Opin. Ther. Targets. 2009;13 (4): 485–496. 98. Dhir M, Montgomery EA, Glöckner SC, Schuebel KE, Hooker CM, Herman JG, Baylin SB, Gearhart SL, Ahuja N. Epigenetic regulation of WNT signaling pathway genes in inflammatory bowel disease (IBD) associated neoplasia. J Gastrointest Surg. 2008;12(10):1745-53. 99. Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993;262: 1731–1734. 100. Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006; 5: 997–1014. 101. Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T,Lawrence Marsh J, Holcombe RF, Waterman ML. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet 2001;28:53–57. 102. Holcombe RF, Marsh JL, Waterman ML, Lin F, Milovanovic T, Truong T. Expression of Wnt ligands and frizzled receptors in colonic mucosa and in colon carcinoma. Mol Pathol. 2002; 55:220– 226. 68 103. Tamura K. Alterations of CDKNZA and mismatch repair deficiency may be important as early events of colorectal tumors associated with ulcerative colitis. Proc AACR 2003;44:1477. 104. Tanaka T, Kohno H, Murakami M, Shimada R, Kagami S. Colitis-related rat colon carcinogenesis induced by 1-hydrozyantraquinone and methylazoxymethanol acetate. Oncol Rep 2000;7:501–508. 105. Suzui M, Ushijima T, Yoshimi N, Nakagama H, Hara A, Sugimura T, Nagao M, Mori H. No involvement of APC gene mutations in ulcerative colitis-associated rat colon carcinogenesis induced by 1-hydrozyanthraquinone and methylazixymethanol acetate. Mol Carcinog 1997;20:389–393. 106. Suzui M, Ushijima T, Dashwood RH, Yoshimi N, Sugimura T, Mori H, Nagao M. Frequent mutations of the rat betacatenin gene in colon cancers induced by methylazoxymethanol acetate plus 1hydrozyanthraquinone. Mol Carcinog 1999;24:232–237. 107. Uthoff SM, Eichenberger MR, Lewis RK, Fox MP, Hamilton CJ, McAuliffe TL, Grimes HL, Falandiuk S. Identification of candidate genes in ulcerative colitis and Crohn’s disease using cDNA array technology. Int. J. Oncology 2001;19:803–810. 108. http://www.stanford.edu/group/nusselab/cgi-bin/wnt/cancer Erişim; 24.04.2012. 109. Zou H, Molina JR, Harrington JJ, Osborn NK, Klatt KK, RomeroY, Burgart LJ, Ahlquist DA. Aberrant methylation of secreted frizzled-related protein genes in esophageal adenocarcinoma and Barrett’s esophagus Int. J. Cancer 2005;116:584–591. 110. Urakami S, Shiina H, Enokida H, Kawakami T, Kawamoto K, Hirata H, Tanaka Y,Kikuno N, Nakagawa M, Igawa M, Dahiya R. Combination Analysis of Hypermethylated Wnt-Antagonist Family Genes as a Novel Epigenetic Biomarker Panel for Bladder Cancer Detection. Clin Cancer Res 2006;12(7): 2109-2116. 111. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem, 1987; 162 (1): 156-159. 112. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods. 2001; 25: 402–408. 113. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nature Protoc, 2008; 3 (6): 1101-1108. 69 114. Schefe JH, Lehmann KE, Buschmann IR, Unger T, Kaiser HF. Quantitative real-time RTPCR data analysis: current concepts and the novel “gene expression’s CT difference” Formula. J Mol Med, 2006; 84: 901–910. 115. Hiltunen MO, Alhonen L, Koistinaho J, Myöhänen S, Pääkkönen M, Marin S, Kosma VM, Jänne J. Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer. 1997;70:644–648. 116. Prasad CP, Mirza S, Sharma G, Prashad R, DattaGupta S, Rath G, Ralhan R. Epigenetic alterations of CDH1 and APC genes: Relationship with activation of Wnt/β-catenin Pathway in invasive ductal carcinoma of breast. Life Sciences 2008; 83: 318–325. 117. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer Nature Reviews Cancer. 2008; 8: 387-398. 118. Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Engeland M van, Toyota M, Tokino T,Hinoda Y, Imai K, Herman JG, Baylin SB. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer Nature Genetıcs 2004; 36 (4):417–422. 119. Dulaimi E, Uzzo RG, GreenbergRE, Al-Saleem T, Cairns P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin Cancer Res, 2004;10:1887- 93. 120. Wang DR, Tang D. Hypermethylated SFRP2 gene in fecal DNA is a high potential biomarker for colorectal cancer noninvasive screening World J Gastroenterol, 2008; 14(4): 524-531. 121. Cheng YY, Yu J, Wong YP, Man EP, To KF, Jin VX, Li J, Tao Q, Sung JJ, Chan FK, Leung WK. Frequent epigenetic inactivation of secreted frizzled-related protein 2 (SFRP2) by promoter methylation in human gastric cancer. British Journal of Cancer. 2007;97: 895 – 901. 122. Takagi H, Sasaki S, Suzuki H, Toyota M, Maruyama R, Nojima M, Yamamoto H, Omata M, Tokıno T, Imai K, Shinomura Y. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma J Gastroenterol 2008; 43:378–389. 123. Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB, Bowers BE, Campbell EL, Colgan SP. An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186(11):6505-14. 70 124. Gonda TA, Kim YI, Salas MC, Gamble MV, Shibata W, Muthupalani S, Sohn KJ, Abrams JA, Fox JG, Wang TC, Tycko B. Folic Acid increases global DNA methylation and reduces inflammation to prevent helicobacter-associated gastric cancer in mice. Gastroenterology. 2012 Apr;142(4):824-833. 125. Nakagawa H, Murata Y, Koyama K, Fujiyama A, Miyoshi Y, Monden M, Akiyama T, Nakamura Y. Identification of a brain-specific APC homologue, APCL, and its interaction with betacatenin. CancerRes. 1998;58(22):5176–5181. 126. van Es JH, Kirkpatrick C, van de Wetering M, Molenaar M, Miles A, Kuipers J, Destree O, Peifer M and Clevers H. Identification of APC2, a homologue of theadenomatous polyposis coli tumour suppressor. Curr Biol. 1999;9 (2):105–108. 127. Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y. APCL, a central nervous system-specific homologue of adenomatous polyposis coli tumor suppressor, binds to p53binding protein 2 and translocates it to the perinucleus. Cancer Res. 2000;60(1):101–105. 128. Bronner MP, Culin C, Reed JC, Furth EE. The bcl-2 protooncogene and the gastrointestinal epithelial tumor progression model. Am J Pathol 1995;146(1):20–26. 71 ÖZGEÇMİŞ AD-SOYAD: Zühal MERT ALTINTAŞ DOĞUM TARİHİ ve YERİ: 03.06.1970 Ankara EĞİTİM DURUMU: 1976-1979: Abdülvahit Sağlam İlkokulu, Ayvalık, BALIKESİR 1979-1981: Sakarya İlkokulu, Ayvalık, BALIKESİR 1981-1983: Ayvalık Lisesi, Ayvalık, BALIKESİR 1983-1987: Mustafa Kemal Lisesi, Yenimahalle, ANKARA 1987-1994: Gazi Üniversitesi Tıp Fakültesi 2004-2006: Mersin Üniversitesi Tıp Fakültesi Tıbbi Biyoloji ve GenetikYüksek Lisans 2006- :Mersin Üniversitesi Tıp Fakültesi Tıbbi Biyoloji ve Genetik Doktora SERTİFİKALAR: 09-15 Kasım 1998: 1.Sağlık Projesi Uyum Eğitimi Sertifikası 15/09/2000: Hemodiyaliz Sorumlu Hekim Sertifikası (Sertifika no:317) DENEYİMLER: Ocak 1995-Ağustos 1996: Merkez Sağlık Ocağı, Taşköprü; Kastamonu Ağustos 1996- Mayıs 1998: 1 Nolu AÇSAP, Karabük Haziran 1998-Ekim 1999: 1 Nolu AÇSAP, Yenimahalle, ANKARA Ekim 1999-Aralık 1999: Acil Müdahale, Türkiye Yüksek İhtisas Hastanesi, Ankara 72 Ocak 2000-Ekim 2001: Hemodiyaliz Ünitesi, Türkiye Yüksek İhtisas Hastanesi, Ankara Mayıs 2001-Ekim 2001: Transplantasyon Koordinasyon Sorumlusu, Türkiye Yüksek İhtisas Hastanesi, Ankara 30 Ekim 2001- 03 Mart 2003 Hemodiyaliz Ünitesi, Devlet Hastanesi, Mersin 03 Mart 2003 –Kasım 2004: Hemodiyaliz Ünitesi, Mersin Üniversitesi Tıp Fakültesi Araştırma ve Uygulama Hastanesi Kasım 2004- : Mersin Üniversitesi Tıp Fakültesi Tıbbi Biyoloji ve Genetik AD. YAYINLAR: 1. Akbas E, Polat S, Karakas-Celik S, Altintas ZM, Yildirim M, Yilgor E. A severely mental and motor retarded boy with monosomy 9pter-->p22 trisomy 10q26-->qter due to paternal reciprocal translocation 46,XY,t(9;10)(p23;q26). Genet Couns. 2011;22(4):417-23. 2. Barlas IO, Sezgin M, Erdal ME, Sahin G, Ankarali HC, Altintas ZM, Türkmen E.Association of (-1,607) 1G/2G polymorphism of matrix metalloproteinase-1 gene with knee osteoarthritis in the Turkish population (knee osteoarthritis and MMPs gene polymorphisms). Rheumatol Int. 2009 Feb;29(4):383-8. 3. Sezgin M, Barlas IO, Ankarali HC, Altintaş ZM, Türkmen E, Gökdoğan T, Sahin G, Erdal ME. Tumour necrosis factor alpha -308G/A gene polymorphism: lack of association with knee osteoarthritis in a Turkish population. Clin Exp Rheumatol. 2008 Sep-Oct;26(5):763-8. 73 4. Sezgin M, Erdal ME, Altintas ZM, Ankarali HC, Barlas IO, Turkmen E, Sahin G. Lack of association polymorphisms of the IL1RN, IL1A, and IL1B genes with knee osteoarthritis in Turkish patients. Clin Invest Med. 2007;30(2):E8692. 74